首页 >
行业动态 > 【Azo-TP-COF】间苯二酚-偶氮二苯胺COF负载FeOOH量子点催化电化学合成氨
【Azo-TP-COF】间苯二酚-偶氮二苯胺COF负载FeOOH量子点催化电化学合成氨
摘要:
Osaka University 的Mio Kondo、Shigeyuki Masaoka和Indian Institute of Science Education and Research的Ramanathan Vaidhyanathan等报道的本篇文章(Chem. Mater. 2024)中报道了一种在环境条件下通过电化学方法合成氨的高效催化剂。研究团队通过精心设计的合成策略,将FeOOH量子点(QDs)均匀分散在一种新型的亚胺连接共价有机框架(IISERP-COF33)上,制备了一种非均相电催化剂(FeOOH@COF)。该催化剂在0.1 M LiClO4水溶液中,于-0.4 V vs可逆氢电极(RHE)的电位下,展现出了77.4 μg h−1 mgcat.−1的氨产率和46.4%的法拉第效率,这一性能超过了目前已知的其他COF和铁基电催化剂。通过一系列材料表征技术,包括粉末X射线衍射(PXRD)、比表面积分析、热重分析(TGA)、傅里叶变换红外光谱(FTIR)、场发射扫描电子显微镜(FESEM)、高分辨透射电子显微镜(HRTEM)和X射线光电子能谱(XPS)等,研究团队详细分析了催化剂的物理化学性质。实验结果表明,FeOOH@COF具有优异的热稳定性、高比表面积和多孔性,这些特性为电催化氮还原反应(NRR)提供了良好的结构基础。此外,通过电化学测试,包括线性扫描伏安法(LSV)和恒电位电解,进一步验证了催化剂的高活性和选择性。本研究为开发新型高效电催化剂提供了重要的理论依据和实验数据,为环境友好的氨合成开辟了新途径。
研究背景:
1) 自19世纪以来,氨(NH3)一直是氮基肥料和制药工业的重要原料。然而,传统的Haber-Bosch氨合成过程存在可持续性挑战,包括巨大的能源消耗和CO2排放。这些问题源于氮的惰性,需要在高温高压(400-450°C和15-25 MPa)下进行反应。
2) 近年来,电催化氮还原反应(ENRR)作为一种有前景且能效高的氨合成方法出现。ENRR提供了在更温和的反应条件下进行NH3生产的潜力,为Haber-Bosch过程提供了一种环保且经济的替代方案。
3) 本文作者通过精心选择构建块,设计了具有战略性放置杂原子的共价有机框架(COF)结构。这种杂原子的引入增强了COF的稳定性、表面积和客体结合能力,使其成为高度通用的催化剂载体。特别是,FeOOH作为一种具有半导体带隙的纳米尺寸量子点(QDs),在COF孔隙中表现出高催化活性。作者合成了一种由FeOOH支持在亚胺连接的共价有机框架(IISERP-COF33)上的非均相电催化剂(FeOOH@COF),在温和条件下实现了高效电催化合成氨。
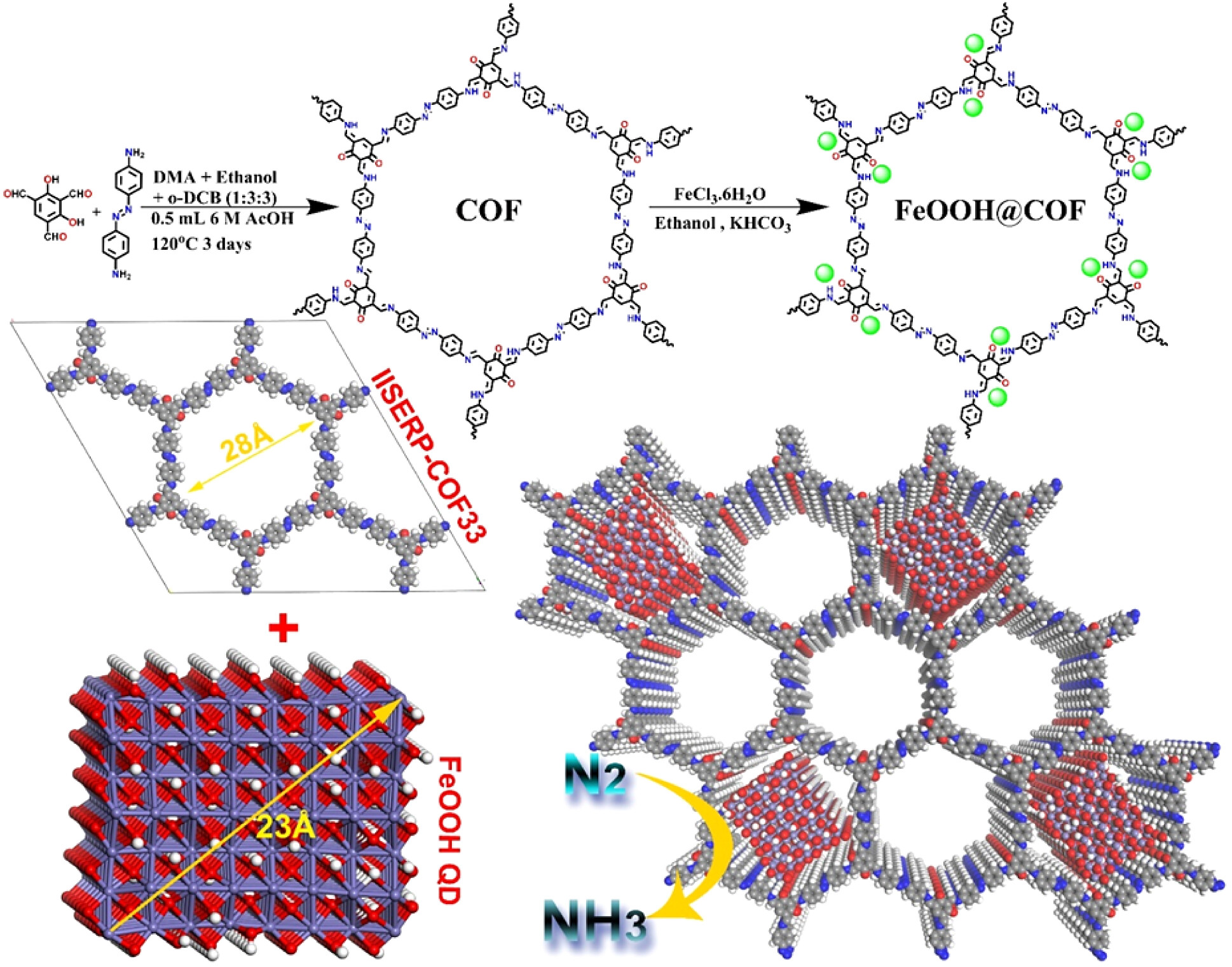
实验部分:
1. IISERP-COF33的合成:
1) 将2,4,6-三甲醛树脂酚(58 mg, 0.3 mmol)和4,4′-偶氮二苯胺(96 mg, 0.45 mmol)溶解在DMA(1.0 mL)、乙醇(3.0 mL)和邻二氯苯(o-DCB, 3.0 mL)的混合溶剂中,搅拌至均匀的绿色溶液。
2) 随后加入6 M 乙酸水溶液(0.5 mL),将混合物转移到Pyrex管中,冷冻后密封并在120 °C下加热3天,然后缓慢冷却至室温。
3) 产物为棕红色沉淀,用甲醇洗涤,得到产率为89%的IISERP-COF33。
2. FeOOH QDs的合成:
1) 将FeCl3·6H2O(1.0 mmol)和KHCO3(3.0 mmol)溶解在乙醇(40 mL)中,搅拌8小时以确保充分反应。
2) 通过离心分离反应产物,并用蒸馏水多次洗涤,最后在40 °C的真空烘箱中干燥以去除残留溶剂和水分。
3. FeOOH@COF的合成:
1) 将FeCl3·6H2O(1.0 mmol)溶解在乙醇(20 mL)中,同时将IISERP-COF33(200 mg)分散在乙醇(20 mL)中,然后将两种溶液混合。
2) 加入KHCO3(3.0 mmol)并继续搅拌8小时,通过离心分离反应产物,并用蒸馏水多次洗涤。
3) 最后,在40 °C的真空烘箱中干燥以去除残留溶剂和水分。
4. 工作电极的制备:
1) 将电催化剂FeOOH@COF(1 mg)加入乙醇(360 μL),然后加入Nafion溶液(10 wt %在乙醇中,40 μL),超声处理至少15分钟以获得均匀的墨水。
2) 将催化剂墨水滴涂在碳布上,负载量为0.5 mg cm−2。
5. 电化学测量:
1) 首先,用Ar气体去除溶解氧和氧基杂质,然后用N2气体吹扫30分钟。
2) 使用带有Nafion 117膜的电化学H型电池进行电催化测试,在室温和常压下进行。
3) 使用涂有催化剂的碳布作为工作电极,Pt线作为对电极,Ag/AgCl(NaCl 3.0 M)作为参比电极。
4) 在0.1 M LiClO4水溶液中进行恒电位电解,电解液体积为9 mL,电解时保持恒定搅拌。
分析测试:
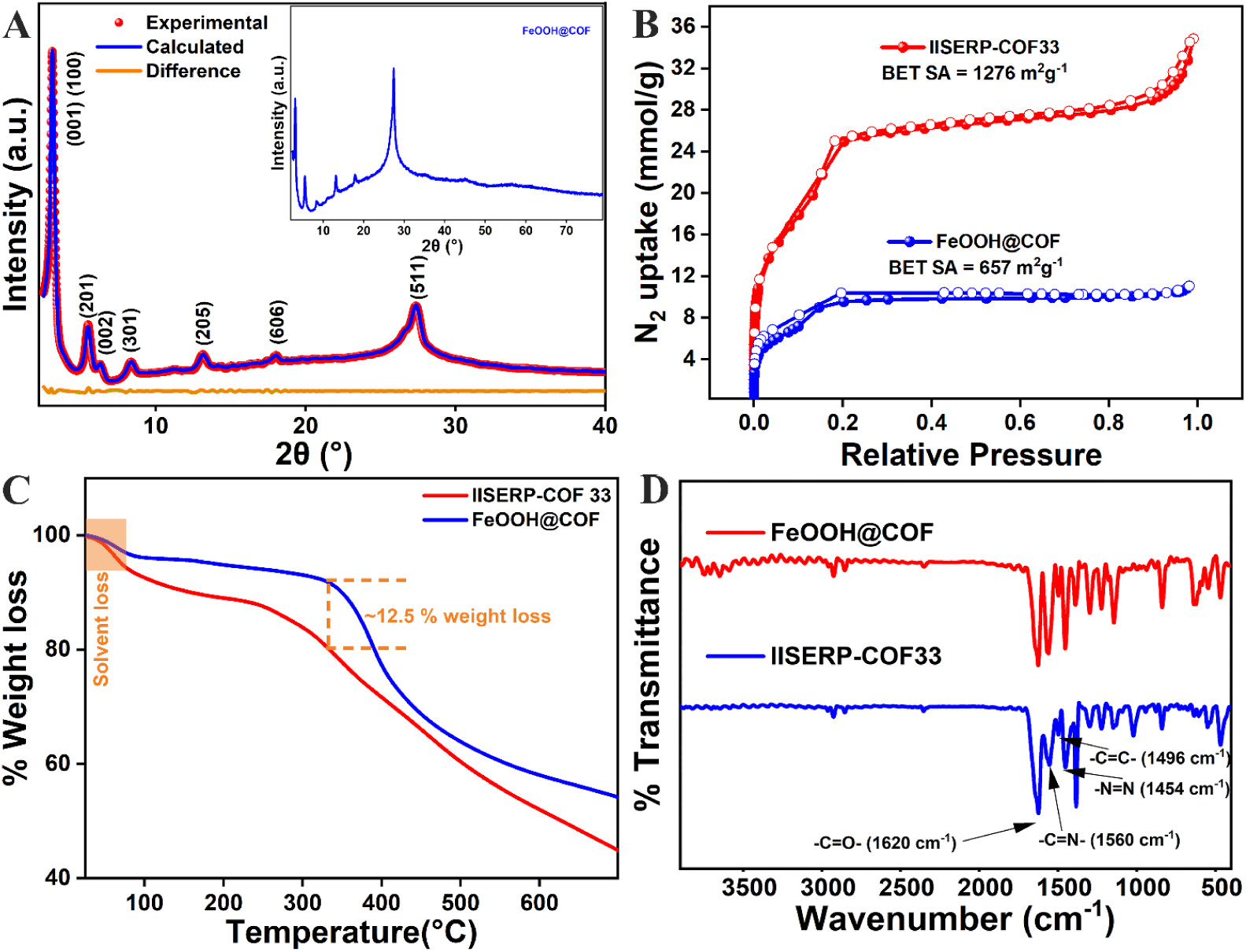
1. 粉末X射线衍射(PXRD)结果:
- IISERP-COF33的PXRD图谱显示了特征峰,表明了其晶体结构。FeOOH@COF的PXRD图谱中未观察到FeOOH的特征峰,表明其为非晶态且尺寸较小。
2. 比表面积和孔隙度分析:
- IISERP-COF33的N2吸附-脱附等温线表现出典型的I型等温线特征,BET比表面积为1276 m2 g−1,Langmuir比表面积为2798 m2 g−1。
- FeOOH@COF的N2吸附-脱附等温线显示饱和N2吸附量降低至10 mmol g−1,表明FeOOH QDs在COF结构中的有效负载。
3. 热重分析(TGA)结果:
- IISERP-COF33和FeOOH@COF的TGA曲线显示,两种材料在空气中均稳定至300 °C,初始质量损失归因于孔隙中溶剂分子的脱附。
4. 傅里叶变换红外光谱(FTIR)结果:
- IISERP-COF33和FeOOH@COF的FTIR谱图显示了特征的C=O(1620 cm−1)、C-N(1560 cm−1)和N=N(1454 cm−1)伸缩振动峰,证实了COF的成功合成和FeOOH的成功负载。
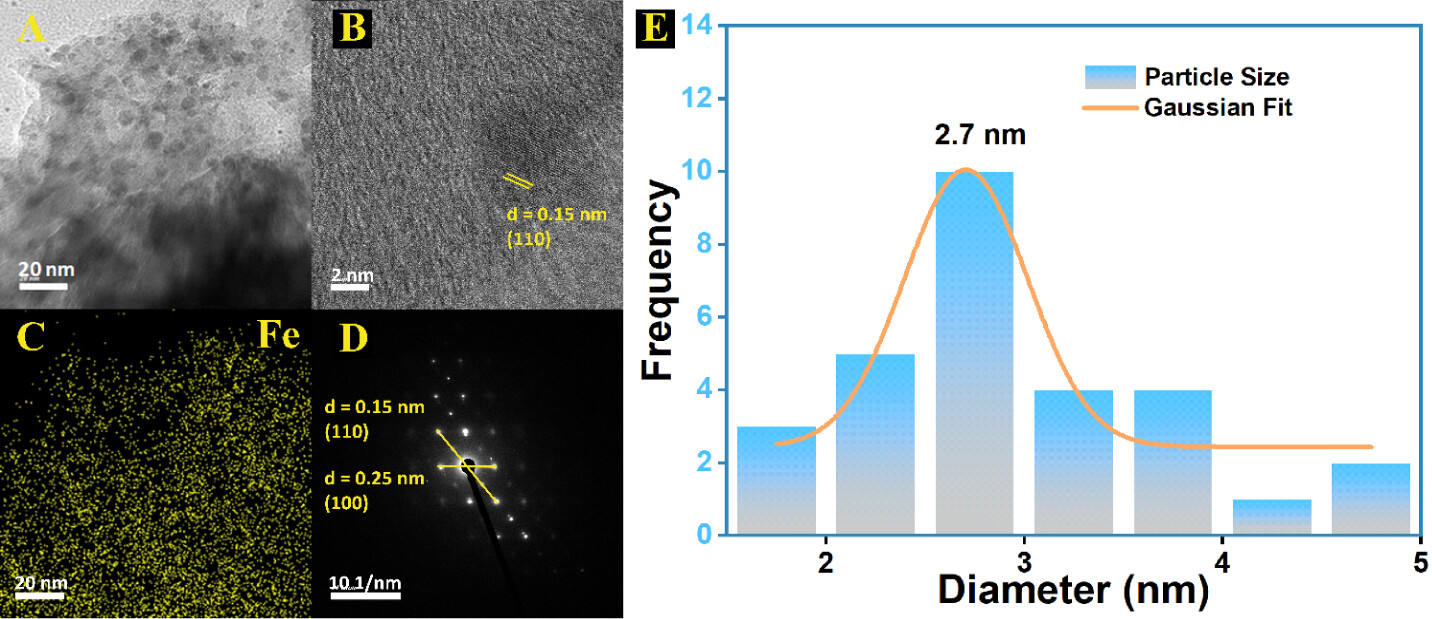
5. 场发射扫描电子显微镜(FESEM)和高分辨透射电子显微镜(HRTEM)结果:
- FESEM图像显示IISERP-COF33和FeOOH@COF具有类似棉絮状的形态。
- HRTEM图像揭示了FeOOH QDs的平均粒径为2.7 nm,且在COF基质中均匀分布。
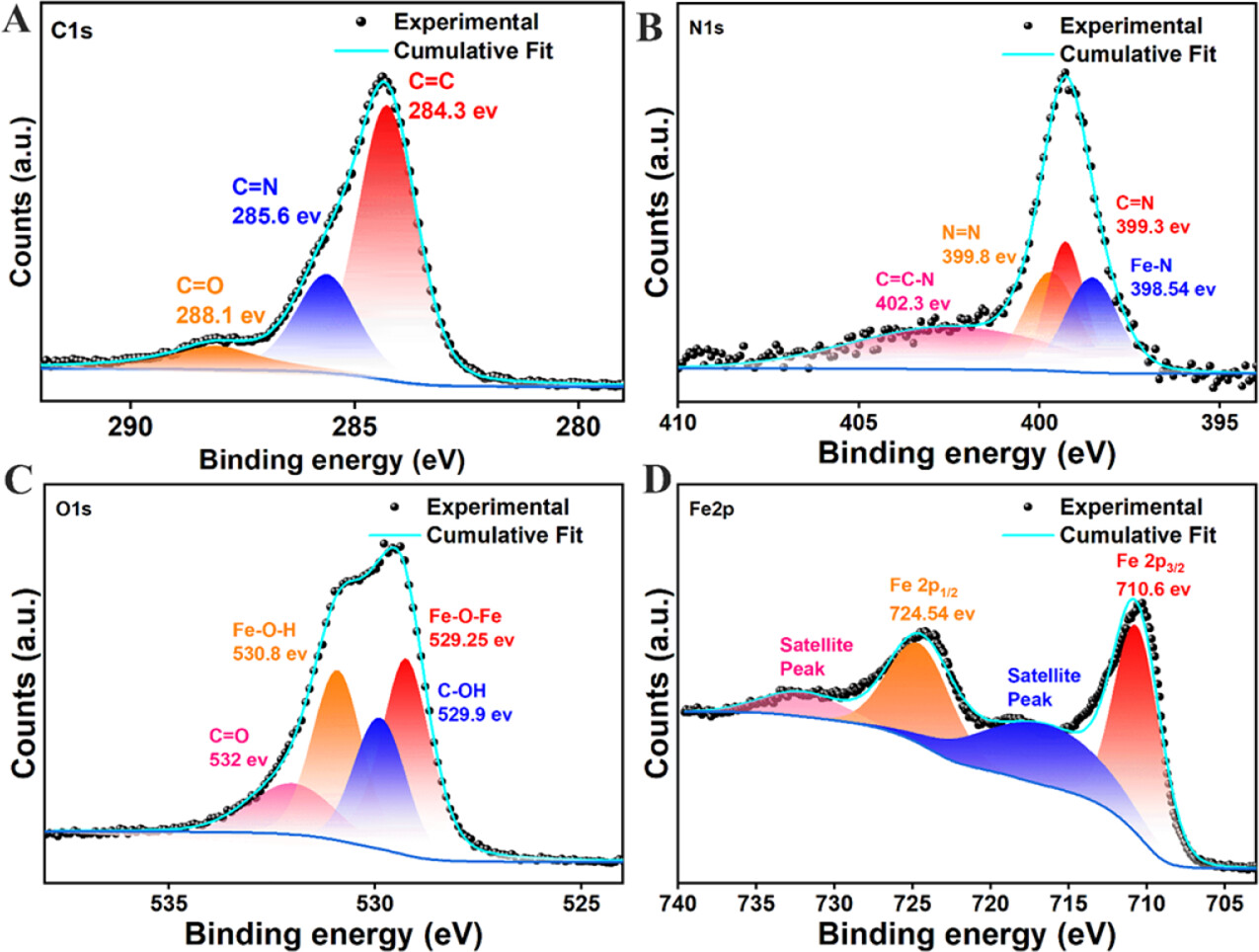
6. X射线光电子能谱(XPS)结果:
- FeOOH@COF的XPS谱图显示了Fe 2p3/2和Fe 2p1/2的特征峰,分别位于710.6 eV和724.54 eV,表明了Fe(III)氧化态的存在。
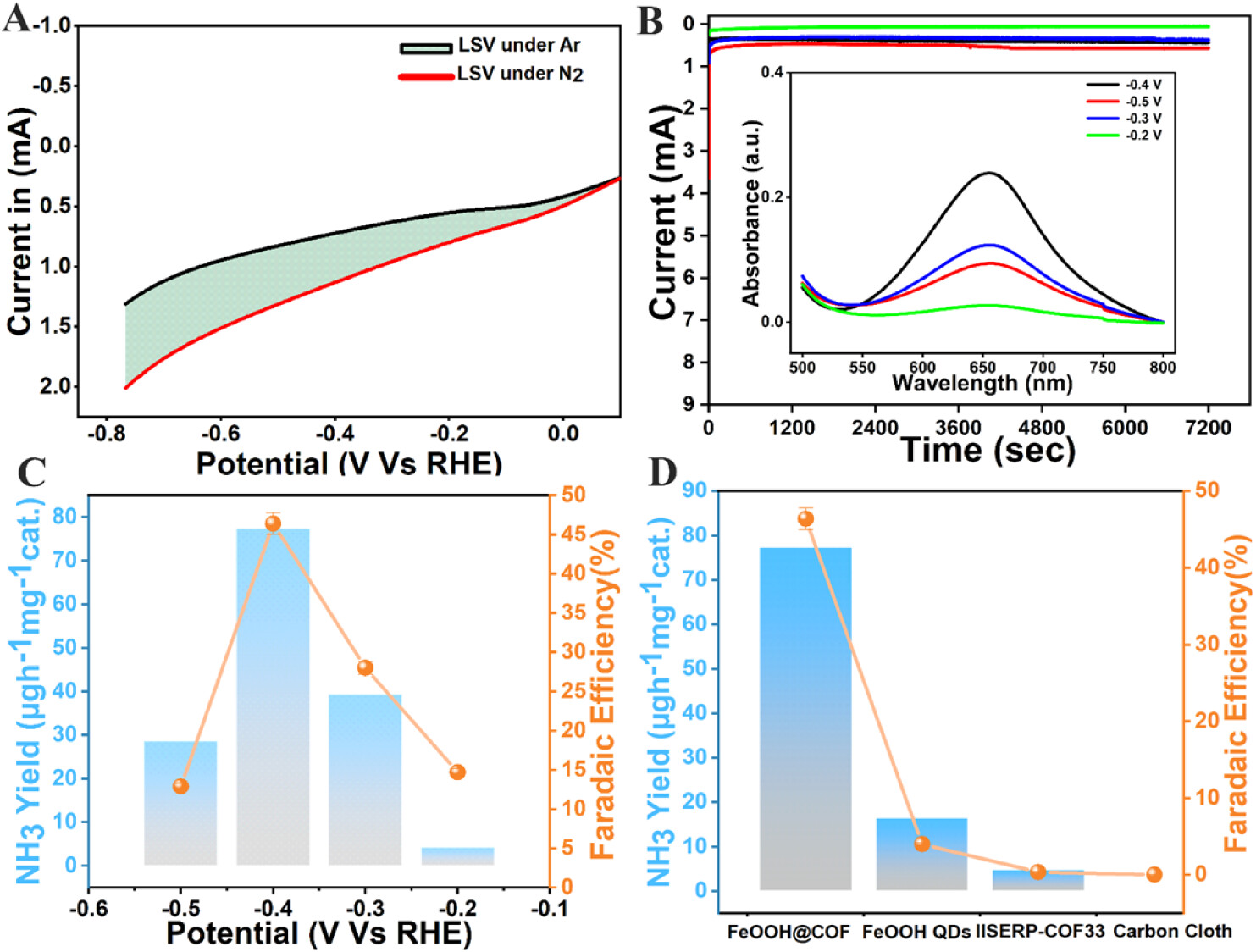
7. 电化学测试结果:
- LSV曲线表明,在N2饱和电解液中FeOOH@COF的电流密度高于Ar饱和电解液,显示出对N2电化学还原的催化活性。
- 恒电位电解结果表明,在-0.4 V vs RHE下,FeOOH@COF的氨产率最高,达到77.4 μg h−1 mgcat.−1,法拉第效率达到46.4%。
8. 氨气的定量分析结果:
- 通过indophenol blue方法测定,FeOOH@COF在-0.4 V vs RHE下电解2小时后,氨的产量为77.4 μg h−1 mgcat.−1。
9. 电化学活性测试结果:
- 时间依赖的电流密度曲线显示,在2小时内FeOOH@COF的电化学氮还原反应(NRR)活性保持稳定。
总结:
本文成功合成了一种新型非均相电催化剂FeOOH@COF,该催化剂在电化学合成氨方面展现出了优异的性能。通过一系列详细的实验和分析测试,证实了FeOOH@COF的结构特征、热稳定性、孔隙性质和电化学性能。这些结果表明,FeOOH@COF是一种高效的电催化剂,为环境友好的氨合成提供了新的可能性。
展望:
本文的科研成果为共价有机框架支持的水稳性量子点的设计提供了系统的方法,为电催化氨合成提供了一种经济的途径。FeOOH@COF为深入的机理研究、多样化的操作探索和规模化尝试提供了一个优秀的系统。将这些经济实惠的系统与可再生能源集成,可以实现氨合成的可持续电催化剂。未来的研究可以探索更多关于COF和FeOOH QDs的合成条件,以进一步提高催化剂的性能和稳定性。此外,研究者可以探索将这种催化剂与其他类型的电化学系统结合,以实现更广泛的应用。
Resorcinol−Azodianiline Covalent Organic Framework Supported FeOOH Quantum Dot-Catalyzed Electrochemical Ammonia Synthesis under Ambient Conditions
文章作者:Pragalbh Shekhar, Kento Kosugi, Himan Dev Singh, Rinku Kushwaha, Deepak Rase, Takumi Matsuzaki, Chitvan Jain, Piyush Singh, Yashraj Singh, Chathakudath Prabhakaran Vinod, Mio Kondo,* Shigeyuki Masaoka,* and Ramanathan Vaidhyanathan*
DOI:10.1021/acs.chemmater.4c00859
文章链接:https://pubs.acs.org/doi/10.1021/acs.chemmater.4c00859
本文为科研用户原创分享上传用于学术宣传交流,具体内容请查阅上述论文,如有错误、侵权等请联系修改、删除。未经允许第三方不得复制转载。


 购销咨询
购销咨询 
