首页 >
行业动态 > 【TAT-COF-1】具有不同取代基的二维氨连接COF结构对六氟化硫吸附分离的理论研究
【TAT-COF-1】具有不同取代基的二维氨连接COF结构对六氟化硫吸附分离的理论研究
摘要:
清源创新实验室Rui Zhao、福州大学侯琳熙和Xiangyu Yin老师等报道的本篇文章(Int J Quantum Chem. 2024;124:e27453)中通过理论计算和模拟研究了二维共价有机框架(COF)材料TAT-COFs-1-AB及其不同功能化基团(SO3H, Et, NH2, OMe, OH, H)对六氟化硫(SF6)的吸附和分离性能。研究发现,低压下SF6的吸附主要取决于与COF框架的相互作用,而高压下主要受孔隙性影响。具有最高孔隙性的TAT-COF-1-AB-H展示了最高的吸附容量8.44 mmol/g(298 K, 100 kPa)。化学功能化被证实可以提高SF6/N2的选择性,其中TAT-COF-1-AB-NH2因其最高的比表面积和强吸附热,展示了最高的选择性。自扩散模拟的结果与GCMC模拟一致。研究强调了吸附容量受取代基和孔隙性的影响,并通过结合能和电荷转移分析证明了SF6对吸附位点的一致偏好。
研究背景:
1. 六氟化硫(SF6)作为一种强效温室气体,在半导体工业尾气净化和回收方面具有显著的经济和环境影响。然而,现有的SF6分离方法如低温蒸馏能耗高、成本高。
2. 物理吸附剂(如沸石、活性炭、多孔聚合物笼、金属-有机框架等)因其经济和环境友好性被考虑用于SF6/N2气体的吸附和分离。
3. 本文创新改进:
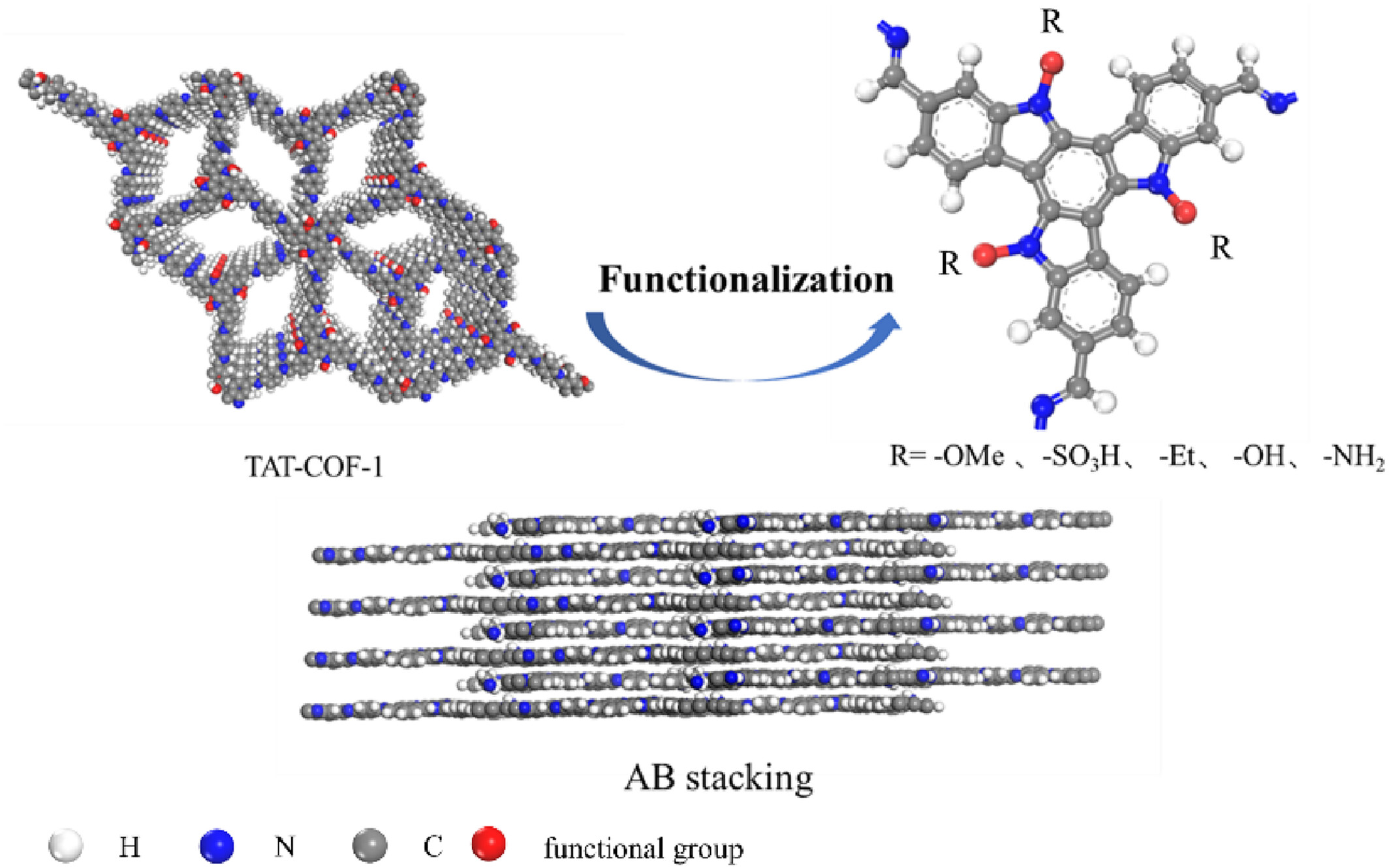
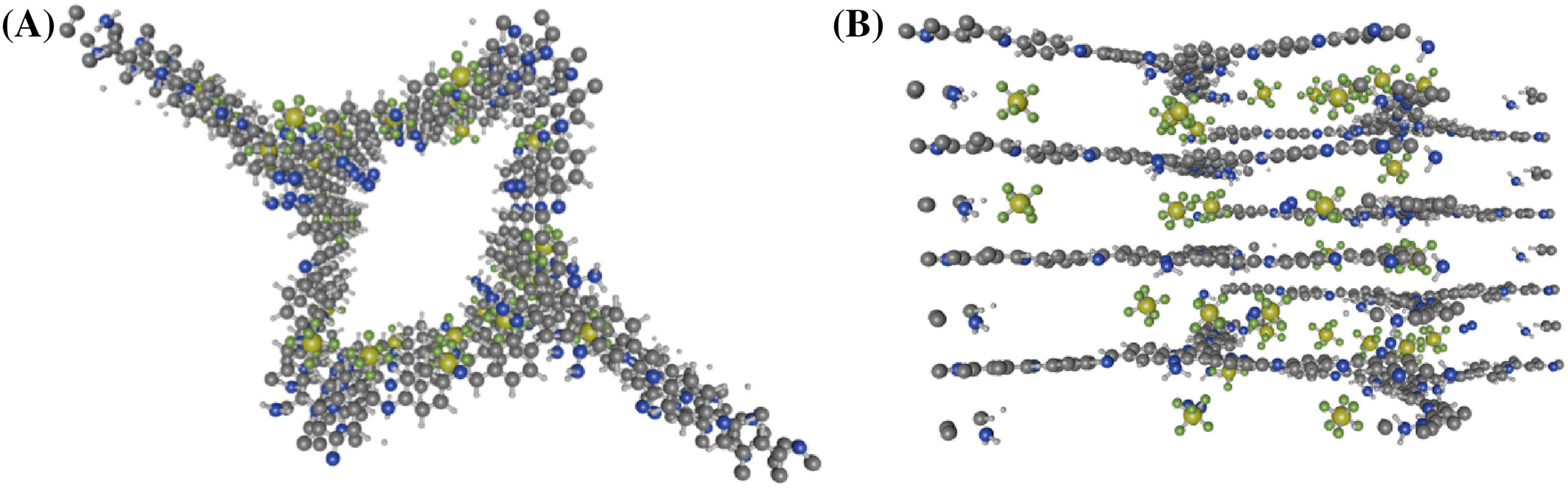
- 提出了使用二维共价有机框架(COF)材料作为吸附剂,该材料具有轻质、热化学稳定性以及可调节的孔径和孔环境。
- 研究了不同功能化基团对COFs吸附SF6性能的影响,特别是对SF6/N2选择性的提升。
- 通过理论计算和模拟,提供了吸附机制和吸附位点的微观理解,为实验设计和改进提供了理论指导。
实验部分:
1. COFs的结构优化和稳定性测试:
- 使用VASP软件包,通过平面波基组和投影增强波(PAW)方法对COFs结构进行优化。
- 采用广义梯度近似(GGA)的Perdew-Burke-Ernzerhof (PBE)泛函,设置平面波截断能为450 eV,自洽场迭代能量截断为1 × 10^(-6) eV。
- 对于每个原子设置力的截断为0.01 eV/Å,不限制对称性,使用1 × 1 × 3的Monkhorst-Pack网格采样布里渊区。
2. 物理特性表征:
- 使用zeo++程序对COFs的孔径、孔隙率、比表面积和孔体积等物理特性进行表征。
- 采用氮气分子作为探针,以1.87 Å的动力学半径进行10000次迭代,以接近实际实验数据。
3. 吸附等温线测定:
- 通过GCMC模拟,在298 K和20-100 kPa条件下,测定了不同取代基团COFs对SF6和N2的吸附等温线。
- 使用RASPA程序进行GCMC模拟,采用Lennard-Jones (LJ)势的σ和ε参数,通过DREIDING力场获得。
4. 选择性测试:
- 在298 K和0-100 kPa条件下,测定了不同取代基团COFs对SF6/N2 (0.1:0.9)二元气体混合物的选择性。
- 采用GCMC模拟,通过计算吸附相和体相中各组分的摩尔分数比来评估分离能力。
5. 自扩散系数计算:
- 通过分子动力学(MD)模拟,在标准NVT系综下计算了SF6和N2在COFs材料中的自扩散系数。
- 使用爱因斯坦关系,通过计算两个客体分子的平均平方位移(MSD)的斜率来计算扩散系数。
6. 吸附位点分析:
- 通过DDEC6电荷计算和电荷密度差(CDD)分析,研究了SF6和N2在不同取代基团COFs上的吸附位点和电子特性。
- 使用Bader电荷分析计算了SF6和N2在COFs上的电荷转移。
分析测试:
1. 凝聚能(Ecoh):
- 所有取代COFs均展现出超过6.75 eV/atom的高凝聚能,表明材料具有良好的稳定性。
2. 比表面积和孔隙特性:
- TAT-COFs-1-AB的比表面积为5732.07 m²/g,孔隙率为0.744,孔径为9 Å,适合SF6吸附。
3. 吸附等温线分析:
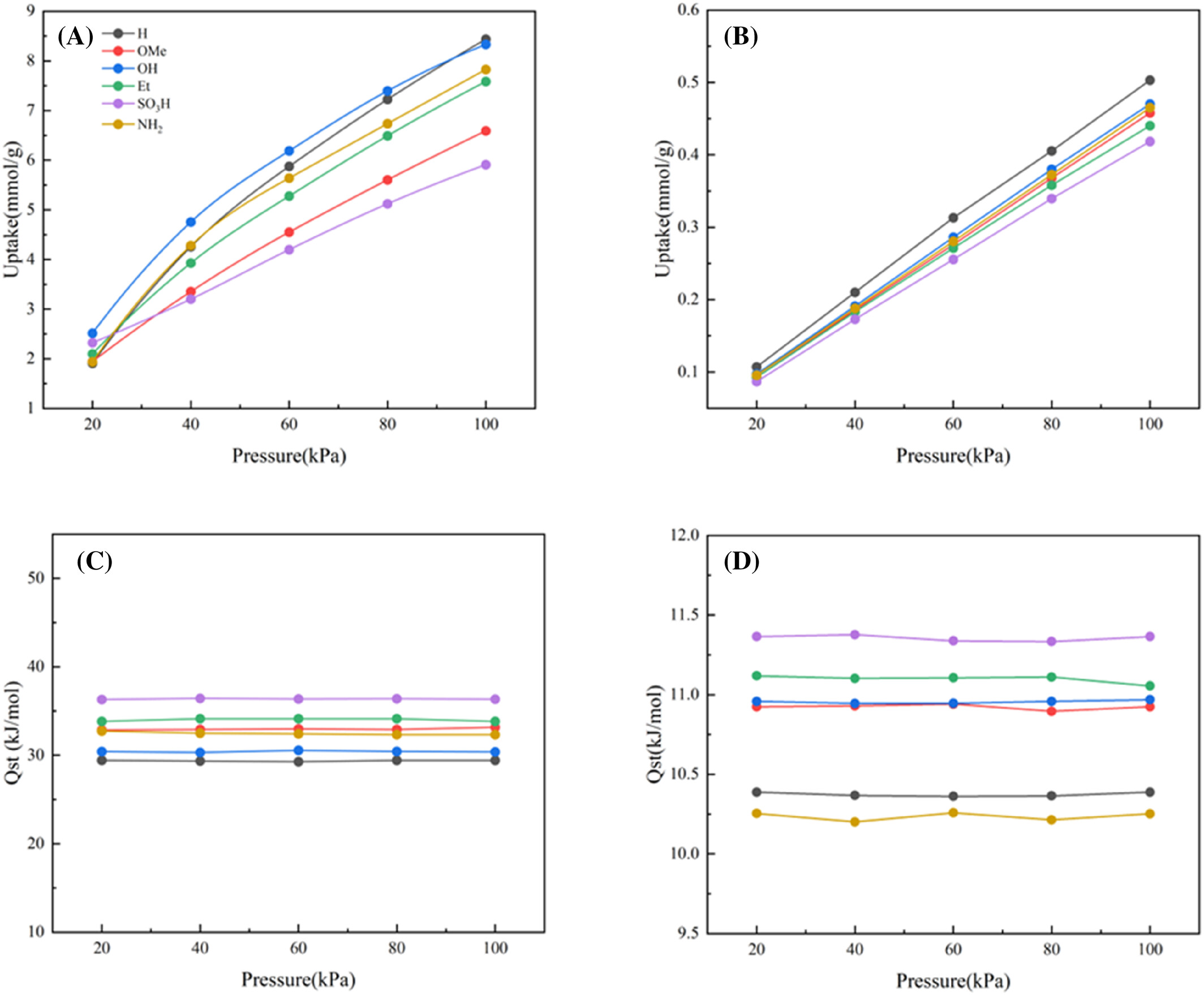
- 在100 kPa下,TAT-COF-1-AB-H对SF6的吸附容量最高,达到8.44 mmol/g。
4. 选择性分析:
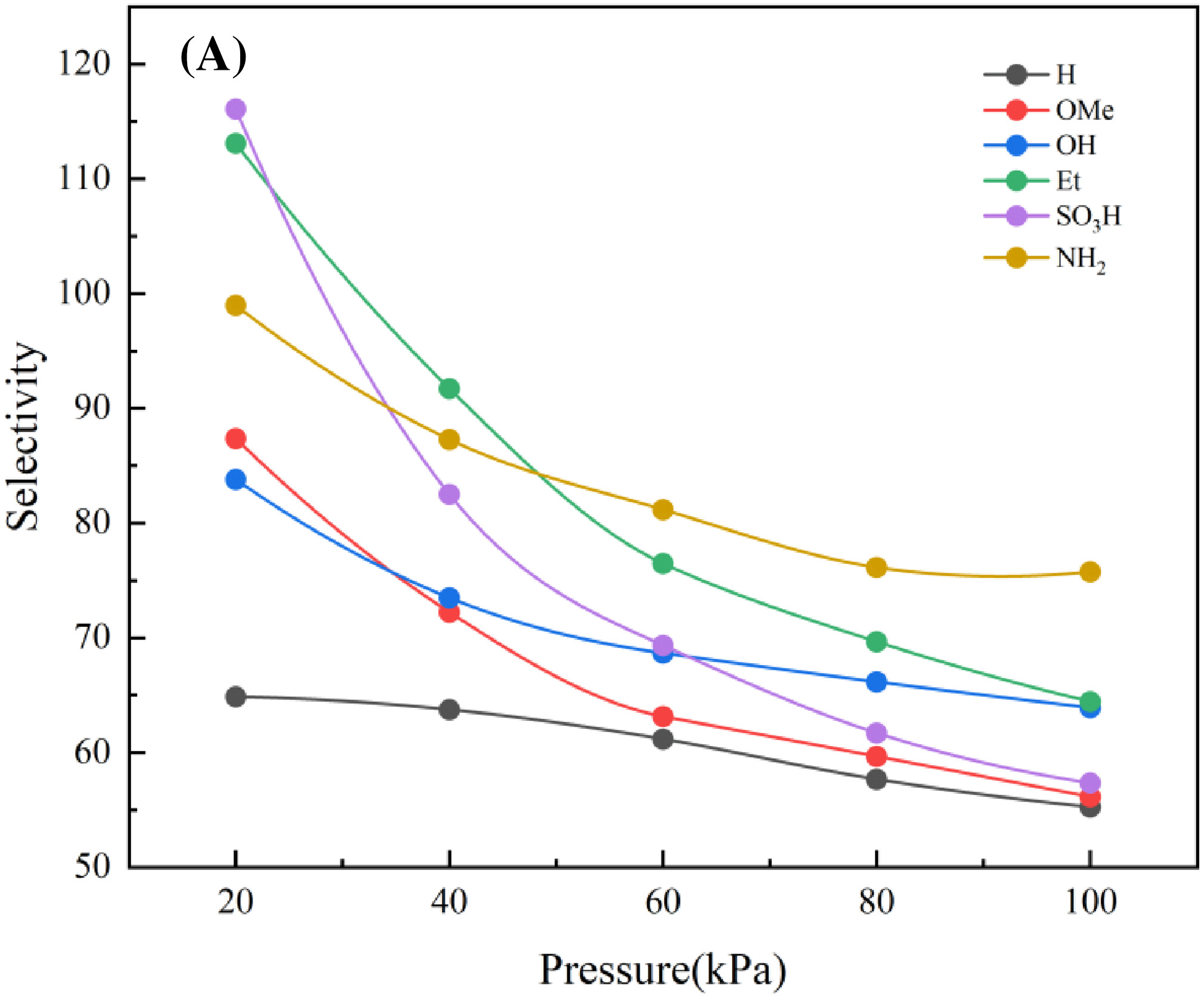
- 在298 K和100 kPa条件下,TAT-COF-1-AB-NH2对SF6/N2的选择性最高,达到75.76。
5. 自扩散系数:
- TAT-COF-1-AB-NH2在SF6/N2 (0.1:0.9)二元气体混合物中的自扩散系数表现最佳,显示了其在净化SF6废气方面的潜力。
6. 吸附位点和电子特性:
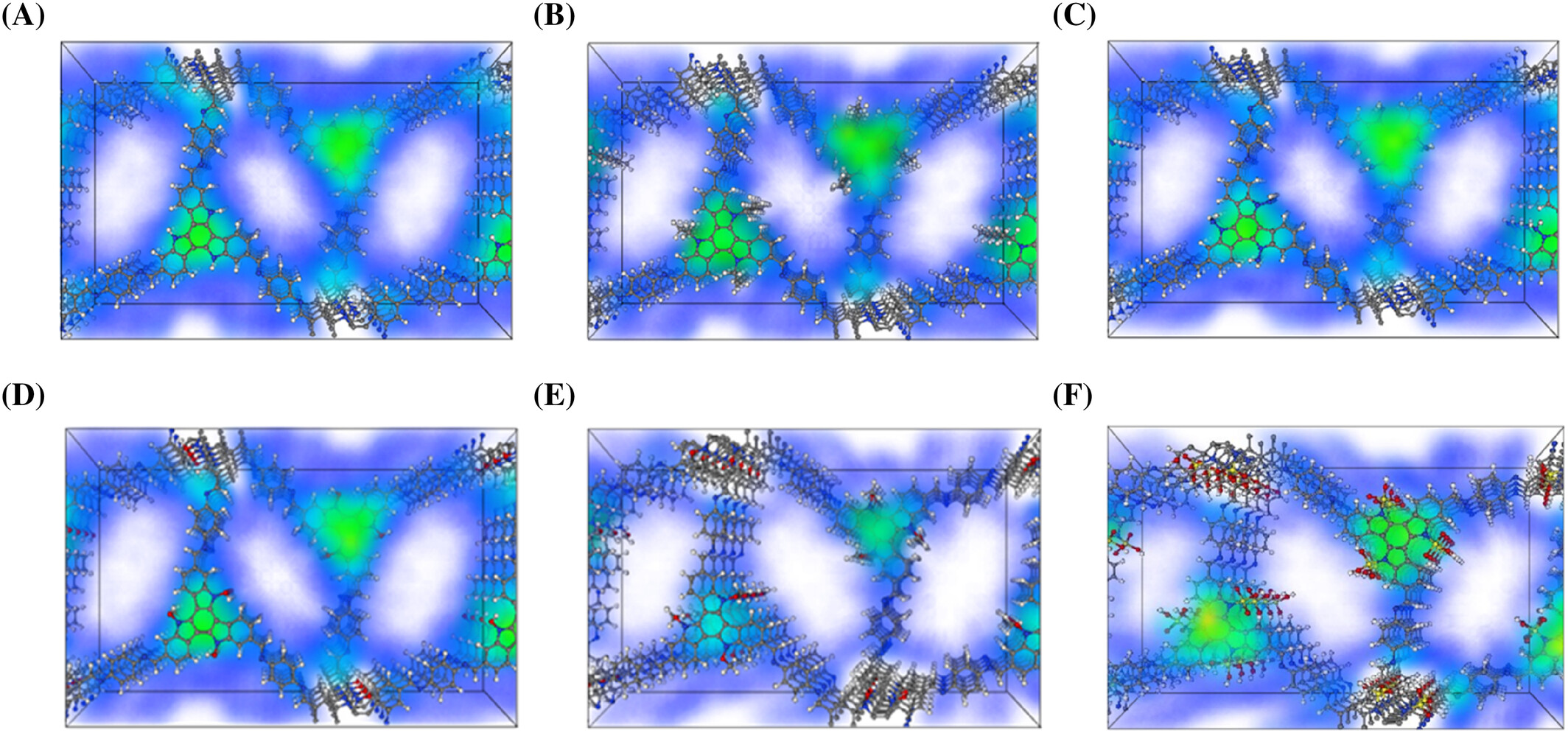
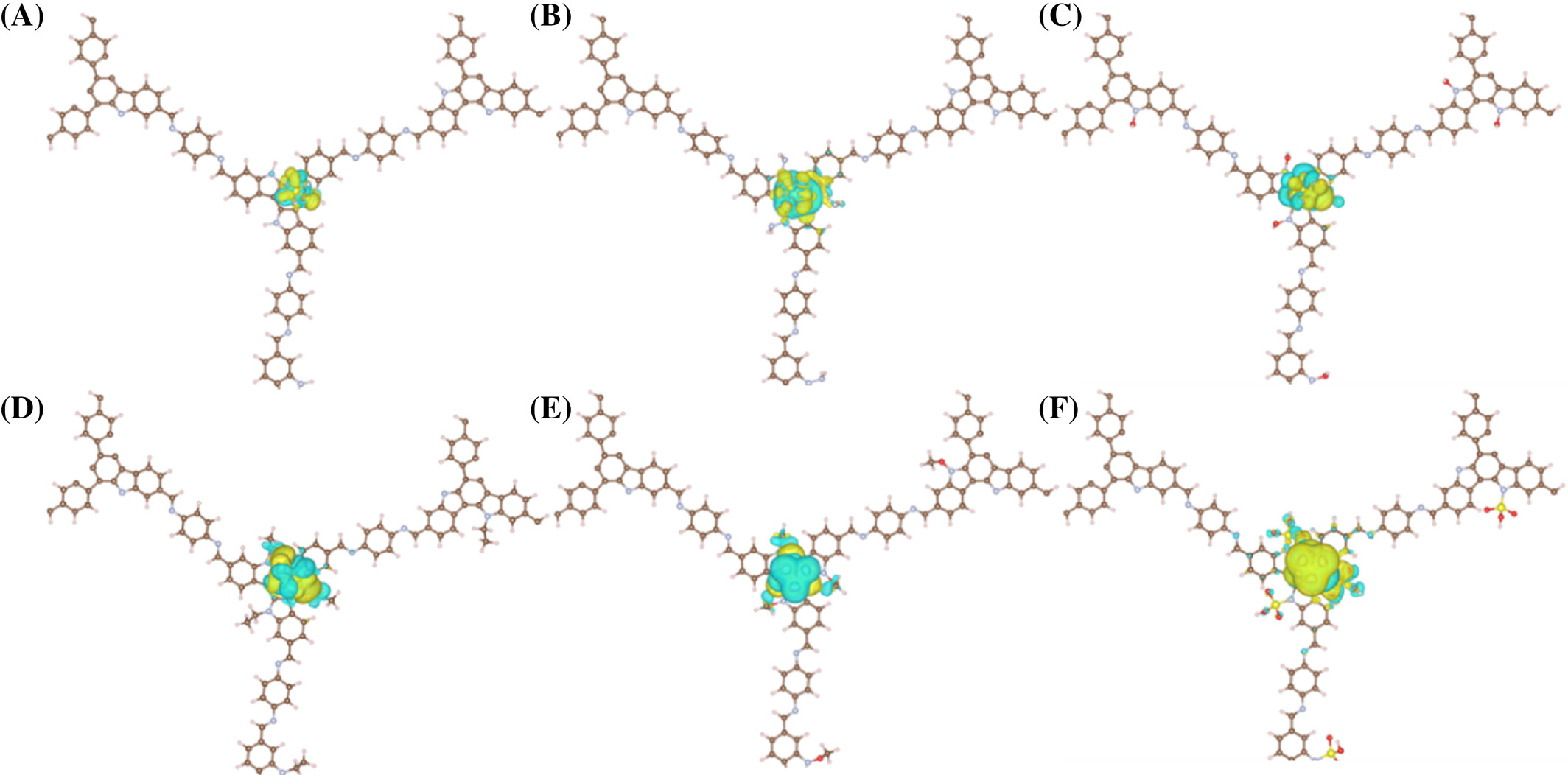
- SF6在AB堆叠的TAT-COFs-1-AB的H位点上表现出最高的吸附能,主要由于强F π相互作用。
- SO3H取代基团与SF6之间的相互作用和电荷转移最强,由于SO3H的高电负性。
6. 其他实验数据:
- 凝聚能的计算结果表明,不同取代基团的COFs均保持了良好的稳定性。
- 比表面积和孔隙特性的详细数据为COFs在SF6/N2分离中的高性能提供了物理基础。
- 吸附等温线和选择性测试结果揭示了不同取代基团对SF6/N2分离性能的显著影响。
- 自扩散系数的计算结果进一步证实了材料在实际应用中的潜在性能。
- 吸附位点和电子特性分析为理解COFs与SF6之间的相互作用提供了深入见解。
总结:
本文通过理论模拟和计算,研究了不同取代基团的二维COF材料对SF6的吸附和分离性能。研究发现,TAT-COF-1-AB-H具有最高的孔隙性和吸附容量,而TAT-COF-1-AB-NH2因其大的比表面积和强吸附热,展现出最高的SF6/N2选择性。自扩散系数的计算结果表明,N2的扩散率远高于SF6,进一步证实了材料的分离潜力。吸附位点和电子特性分析揭示了SF6在COFs上的吸附偏好和机制。
展望:
本文的研究为SF6的吸附和分离提供了新的材料选择和理论依据。未来的工作可以在以下几个方面进行:
1. 基于理论模拟结果,合成具有最佳性能的COF材料,并进行实验验证。
2. 进一步研究不同功能化基团对COFs稳定性和电子特性的影响。
3. 探索COFs在实际工业环境中对SF6/N2混合气体的吸附和分离性能。
Two-dimensional ammonia-linked COF structures with different substituents for the adsorption and separation of sulfur hexafluoride: A theoretical study
文章作者:Kun Shen, Junjie Ning, Rui Zhao, Kunqi Gao, Xiangyu Yin, Linxi Hou
DOI:10.1002/qua.27453
文章链接:https://onlinelibrary.wiley.com/doi/epdf/10.1002/qua.27453
本文为科研用户原创分享上传用于学术宣传交流,具体内容请查阅上述论文,如有错误、侵权等请联系修改、删除。未经允许第三方不得复制转载。


 购销咨询
购销咨询 
