首页 >
行业动态 > 【MOF-808】金属有机框架中的锆位点 - 羟基对介导过氧物种生成,实现芳香胺的高效串联氧化缩合
【MOF-808】金属有机框架中的锆位点 - 羟基对介导过氧物种生成,实现芳香胺的高效串联氧化缩合
摘要
国家纳米科学中心李国栋老师团队报道研究的针对芳香胺选择性氧化为氧化偶氮芳烃的反应路径复杂、选择性调控难问题,本研究以均苯三甲酸锆基金属有机框架 MOF-808 为模型催化剂,构建路易斯酸性锆位点与端羟基的双活性位点体系。该体系通过氢键活化过氧化氢生成非自由基型 Zr-OOH 活性物种,实现苯胺经中间体串联氧化缩合生成氧化偶氮苯的非自由基反应。25℃下该催化剂实现苯胺近乎完全转化,对氧化偶氮苯选择性达 97.1%,15 次连续循环仍保持良好稳定性,性能远优于 ZrO₂和 Zr (OH)₄。研究实现氧化偶氮苯克级合成,验证了体系对 22 种苯胺衍生物均相缩合、6 种非均相缩合的普适性,且该双位点策略可拓展至其他锆 / 铪基金属有机框架,为过氧化氢活化及选择性氧化反应开辟新途径。
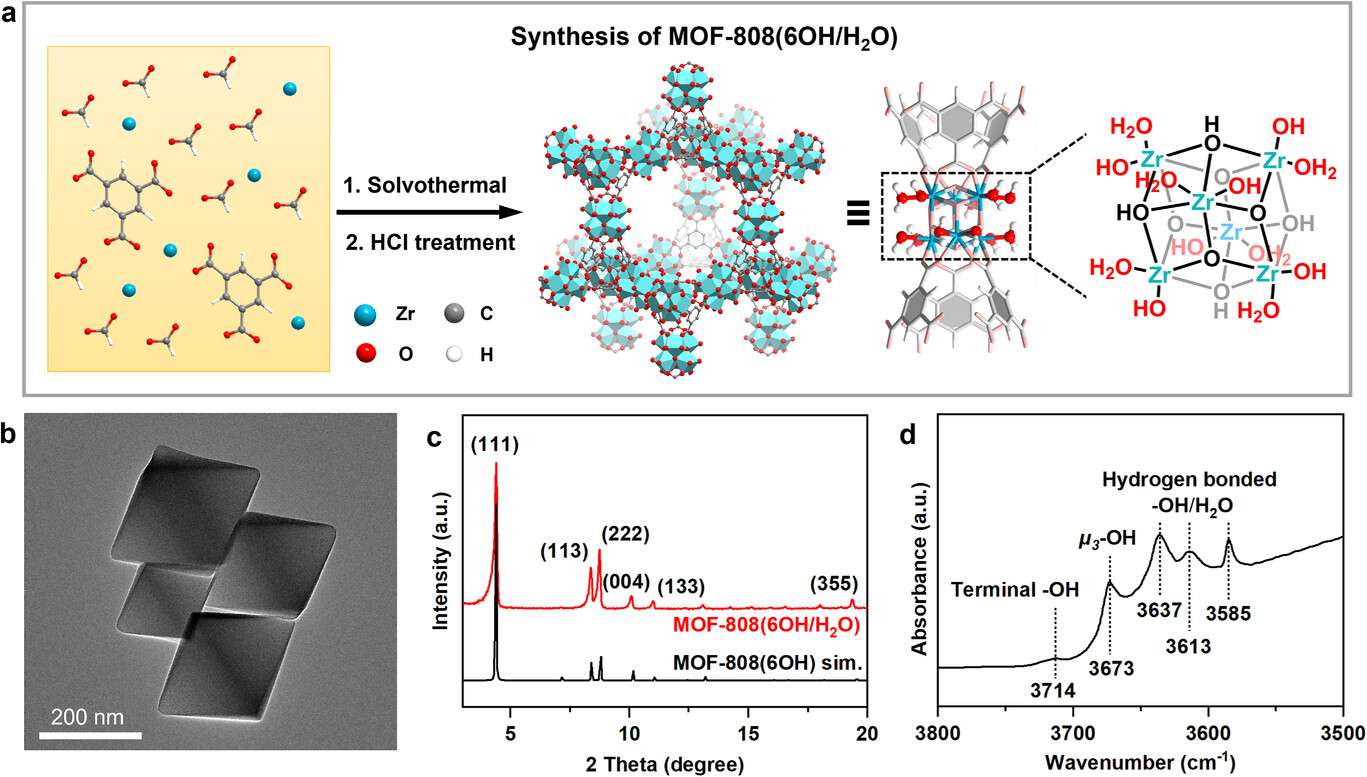
研究背景
1. 行业核心问题:
氧化偶氮芳烃在染料、医药等领域应用广泛,由芳香胺选择性氧化制备该产物是研究热点,但反应路径复杂、副产物多,选择性调控难度大;传统氧化剂易产生有害废弃物,过氧化氢作为绿色氧化剂,现有催化体系对其活化效率低,且酸性环境易导致氧化偶氮苯重排。
2. 现有解决方案局限:
酸催化剂催化苯胺氧化高转化率但氧化偶氮苯收率低于 10%;含添加剂的非均相催化剂分离纯化复杂,限制工业应用;无添加剂非均相催化剂存在选择性或收率短板;锆基催化剂虽有潜力,但活性中心认知模糊、过氧化氢活化机制存争议,端羟基作用未被重视。
3. 本文创新改进:
利用 MOF-808 的开放锆位点和可调孔结构,构建锆位点与相邻端羟基的双位点协同体系,首次明确端羟基的关键作用;通过非自由基路径生成 Zr-OOH 活性物种,突破传统锆基催化剂的自由基反应局限;系统验证该策略对多种底物和金属有机框架的普适性,为绿色氧化剂活化提供新的催化设计思路。
实验部分
1. MOF-808 (6OH/H₂O) 合成:
以八水合氧氯化锆、均苯三甲酸为前驱体,DMF 为溶剂、甲酸为调节剂,100℃溶剂热制备初始 MOF-808;经 1mol/L 盐酸 90℃搅拌 48h 脱除甲酸根,得到组成为 Zr₆(μ₃-O)₄(μ₃-OH)₄(BTC)₁.97 (FA)₀.12 (OH)₅.97 (H₂O)₅.97 的目标催化剂,成功构建端羟基位点。
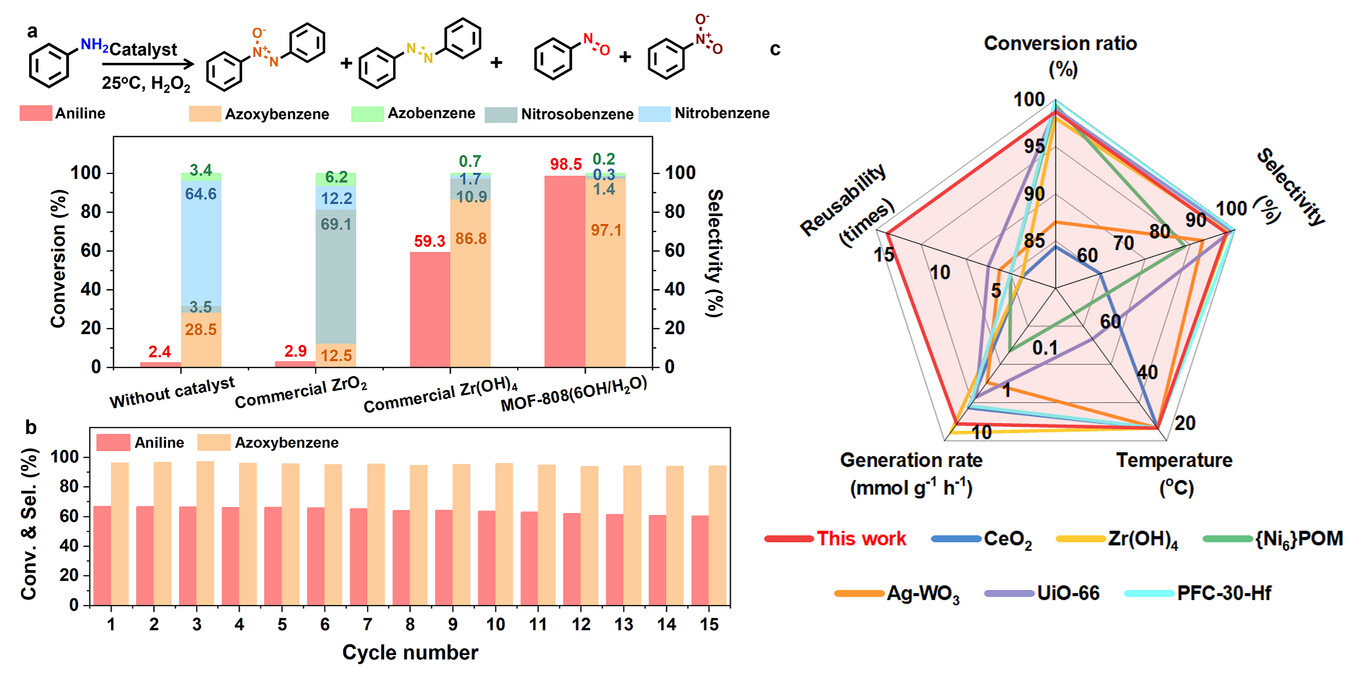
2. 催化性能测试:
25℃下以乙醇为溶剂,1mmol 苯胺、20mg 催化剂、220μL 30% 过氧化氢反应 1.5h,MOF-808 (6OH/H₂O) 实现 98.5% 转化率、97.1% 氧化偶氮苯选择性,生成速率 16.0 mmol・g⁻¹・h⁻¹,活性为 Zr (OH)₄的 4 倍;升温会提升转化率但降低选择性。
3. 稳定性与规模化实验:
在苯胺转化率约 65% 和近 100% 条件下,15 次循环催化的转化率和选择性均无明显变化,锆溶出量低于 0.014%;20mmol 苯胺克级反应实现 96.6% 转化率、93.3% 选择性,柱层析后氧化偶氮苯分离收率 88.9%。
4. 普适性实验:
22 种含吸 / 供电子基的苯胺衍生物、杂环芳胺及芳香二胺均实现高转化率(多 > 95%)和高选择性(多 > 90%);6 种不同芳香胺的非均相缩合也成功制备目标产物,证实广谱官能团耐受性。
5. 催化剂拓展实验:
在 NU-1000、DUT-67 等锆基金属有机框架,及铪基 MOF-808、NU-1000 中构建同类金属位点 - 端羟基对,所有催化剂均表现出优异催化性能,验证策略通用适用性。
实验突破:首次实现非自由基型 Zr-OOH 介导的芳香胺高效氧化缩合,解决传统体系选择性低、稳定性差问题;实现克级合成和 15 次稳定循环,突破实验室小试局限;验证了双位点体系在多种催化剂和底物中的普适性。
分析测试
1. 形貌与晶相:
TEM 显示 MOF-808 (6OH/H₂O) 为八面体,粒径 184±42nm;XRD 特征峰与模拟值一致,晶相纯度高,循环后仅峰强轻微下降,晶相稳定。
2. 孔结构与表面性质:
N₂吸附 - 脱附测试显示其比表面积 2405m²・g⁻¹,平均孔径 1.75nm,与截角四面体孔笼结构一致;循环后孔结构参数仅轻微降低,多孔结构保持良好。
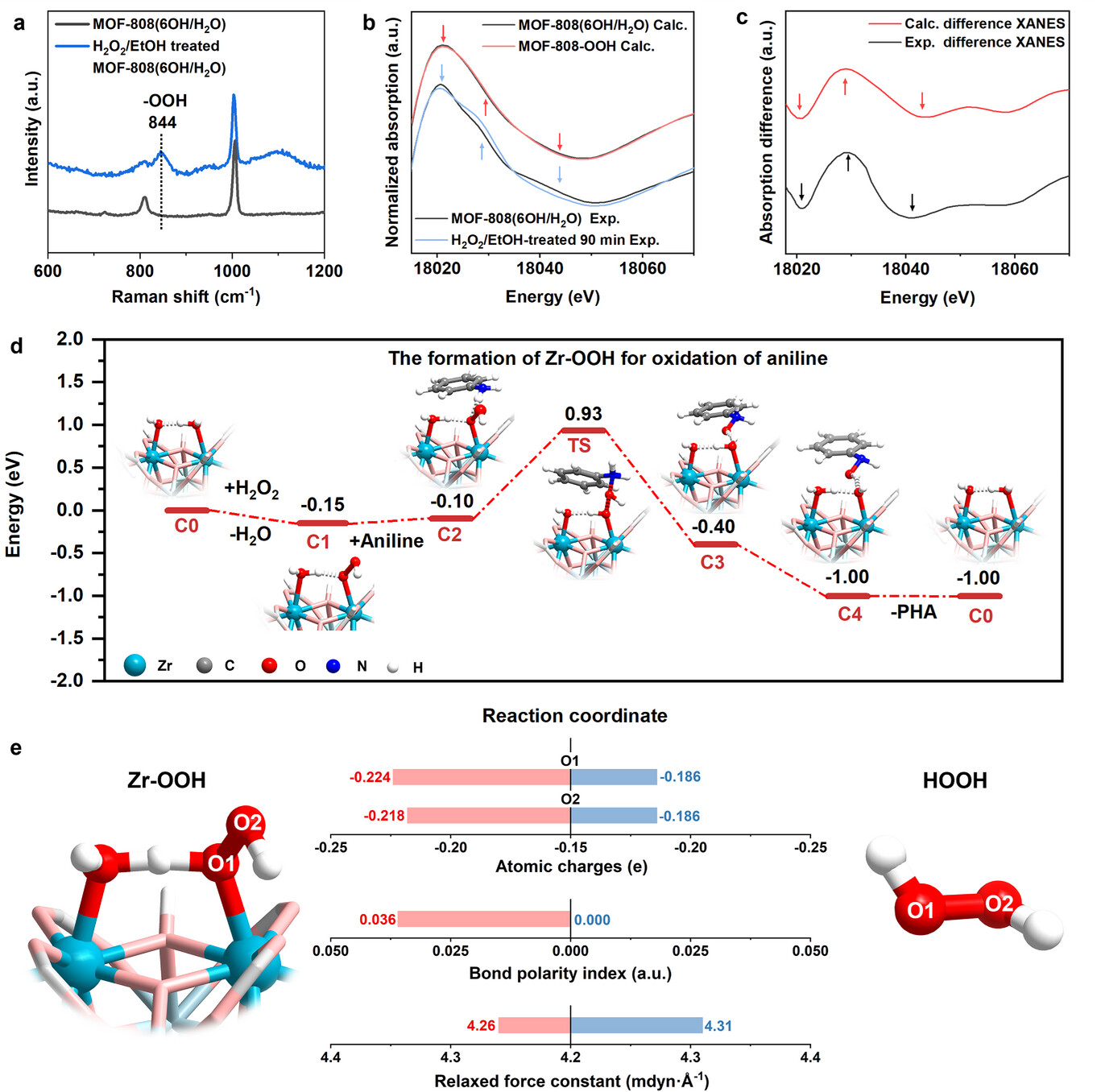
3. 光谱与能谱:
DRIFTS 检测到端羟基、μ₃-OH 及氢键结合的 OH/H₂O 特征峰,证实端羟基成功构建;拉曼光谱在 844cm⁻¹ 出现 Zr-OOH 的 O-O 键特征峰,证实过氧物种生成;XAFS 和 XPS 证实 Zr-OOH 物种形成,且反应后锆氧化态、端羟基位点基本不变。
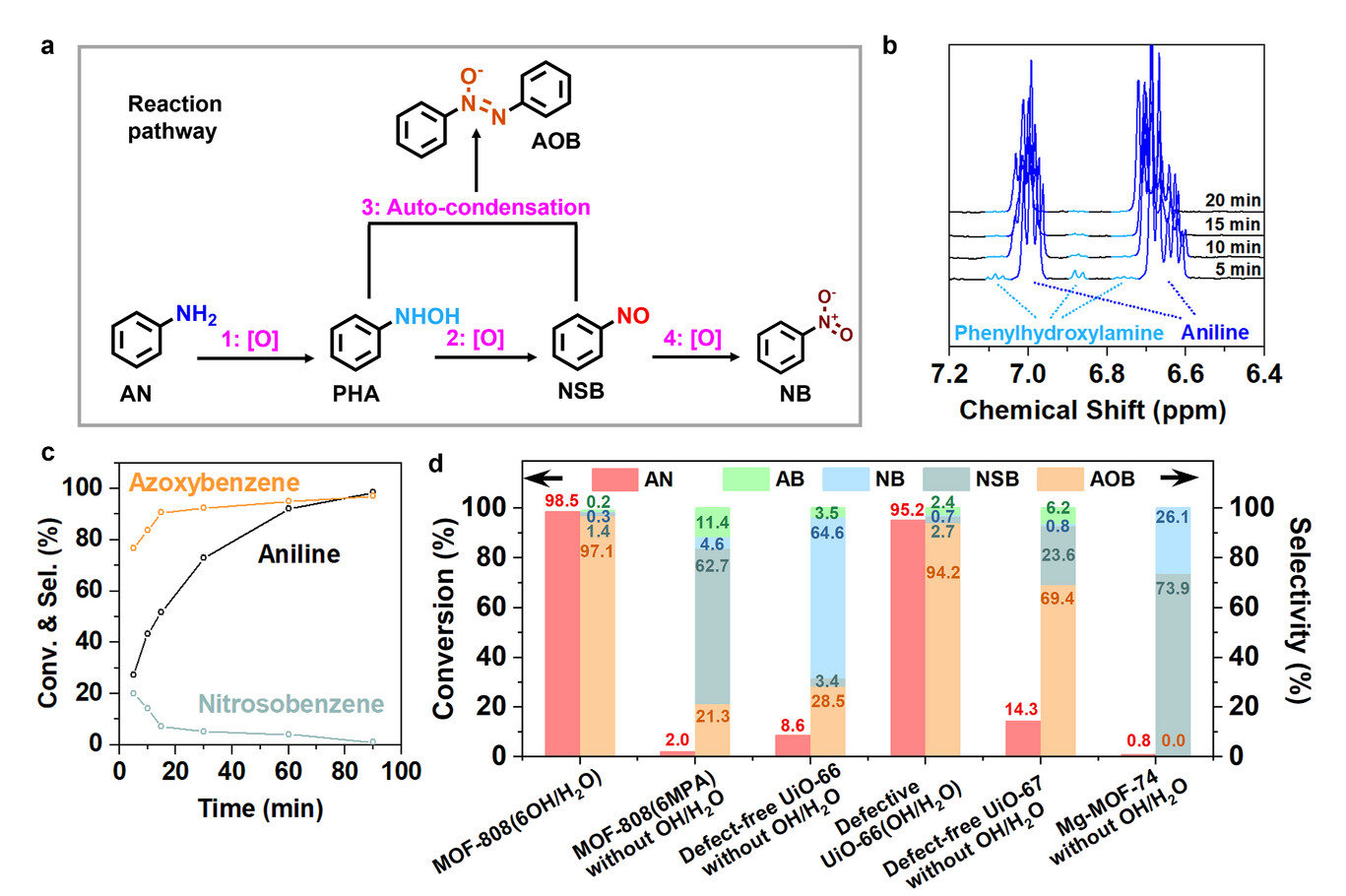
4. 自由基与反应跟踪:
EPR 未检测到羟基自由基,仅见微弱超氧阴离子信号,加入自由基清除剂后催化性能无变化,证实非自由基路径;¹H NMR 跟踪发现 N - 苯基羟胺快速消长,亚硝基苯选择性先升后降,氧化偶氮苯选择性持续升高,明确中间体转化规律。
测试结果揭示:盐酸后处理可有效构建 MOF-808 端羟基位点,形成锆位点 - 端羟基双活性中心;该体系通过氢键活化过氧化氢生成非自由基 Zr-OOH,是催化反应核心活性物种;MOF-808 的多孔结构和稳定晶相为催化反应提供良好传质环境和结构基础。
机理分析
1. 过氧化氢活化机理:
锆位点吸附过氧化氢,相邻端羟基通过氢键形成协同作用,促进氢原子转移生成非自由基 Zr-OOH(能量 - 0.15eV),无需化学键均裂产生自由基;Zr-OOH 的 O-O 键极性更高、松弛力常数更低(4.26mdyn・Å⁻¹),相比过氧化氢(4.31mdyn・Å⁻¹)反应活性更强。
2. 氧化缩合反应机理:
反应遵循非自由基串联路径,Zr-OOH 通过亲核进攻氧化苯胺生成 N - 苯基羟胺,该步骤决速步为 Zr-OOH 的 O-O 键解离(能垒 1.03eV),远低于苯胺与自由过氧化氢的反应能垒(1.54eV);N - 苯基羟胺快速氧化为亚硝基苯,二者自发缩合生成氧化偶氮苯,缩合速率远快于副产物生成,实现高选择性。
3. 双位点协同机理:
锆位点为路易斯酸中心,负责吸附活化底物和过氧化氢;端羟基作为辅助位点,通过氢键调控过氧化氢吸附构型、降低活化能垒,促进 Zr-OOH 生成;二者空间邻近性是关键,端羟基被完全封闭后催化剂几乎丧失活性。
总结
1. 成功在 MOF-808 中构建路易斯酸性锆位点 - 端羟基双活性位点体系,明确了端羟基在过氧化氢活化中的关键作用,首次实现非自由基型 Zr-OOH 物种的可控生成。
2. 该催化体系在 25℃温和条件下实现苯胺的高效选择性氧化缩合,转化率和选择性均优于现有锆基催化剂,且具有优异的循环稳定性和克级合成能力,为绿色氧化合成提供了新的高效催化剂。
3. 系统验证了催化体系的普适性,不仅适用于 22 种苯胺衍生物的均相缩合和 6 种非均相缩合,还可拓展至其他锆 / 铪基金属有机框架,证实了位点协同策略的通用性。
4. 结合实验表征和理论计算,阐明了过氧化氢的活化机制和苯胺氧化缩合的非自由基反应路径,为多步选择性氧化反应的催化设计提供了理论指导。
文章标题:Pairs of Zirconium Site and Hydroxyl in MOFs EnableGenerating Peroxo Species for Efficiently Tandem OxidativeCondensation of Aromatic Amines
文章作者:Zhiyong Ban, Qing Feng, Caoyu Yang, Siyang Li, Hanlin Liu, Haoyu Zhang, Lulu Zuo, Xinwei Li, Long Jiao, Guodong Li
DOI:10.1002/adma.72877
文章链接:https://doi.org/10.1002/adma.72877
本文为科研用户原创分享,用于学术宣传交流,具体细节请查阅原文。如有错误、侵权,请联系修改删除,未经允许不得复制转载。


 购销咨询
购销咨询 
