首页 >
行业动态 > 【Ce-UiO-66-NH2】光诱导瞬态开放Ce(III)位点实现MOF中CO₂的可逆结合与还原
【Ce-UiO-66-NH2】光诱导瞬态开放Ce(III)位点实现MOF中CO₂的可逆结合与还原
摘要:
北大杨四海与曼彻斯特大学Martin Schröder教授团队在(J. Am. Chem. Soc., 2026, DOI: 10.1021/jacs.5c20721),报道了基于铈的金属有机框架材料 Ce-UiO-66-NH2,在光照下能够可逆地结合 CO2,并在无需牺牲剂的情况下,在水中将 CO2 光催化还原为 CO。研究发现,该材料的 CO 生成速率高达 126 μmol・g⁻¹・h⁻¹,且选择性为 100%,性能优于其无胺基类似物 Ce-UiO-66 及目前报道的基准催化剂。通过原位红外、X 射线吸收、电子顺磁共振和瞬态吸收光谱等表征手段,揭示了光激发诱导配体到金属的电荷转移(LMCT),从而生成瞬态开放的 Ce (III) 位点,该位点以 μ-(η¹-O)(η¹-C) 模式结合 CO2。这种结合是可逆的,并能激活 CO2 进行后续的光还原生成 CO。该工作为设计能够从 CO2 合成燃料的光催化剂提供了新的思路。
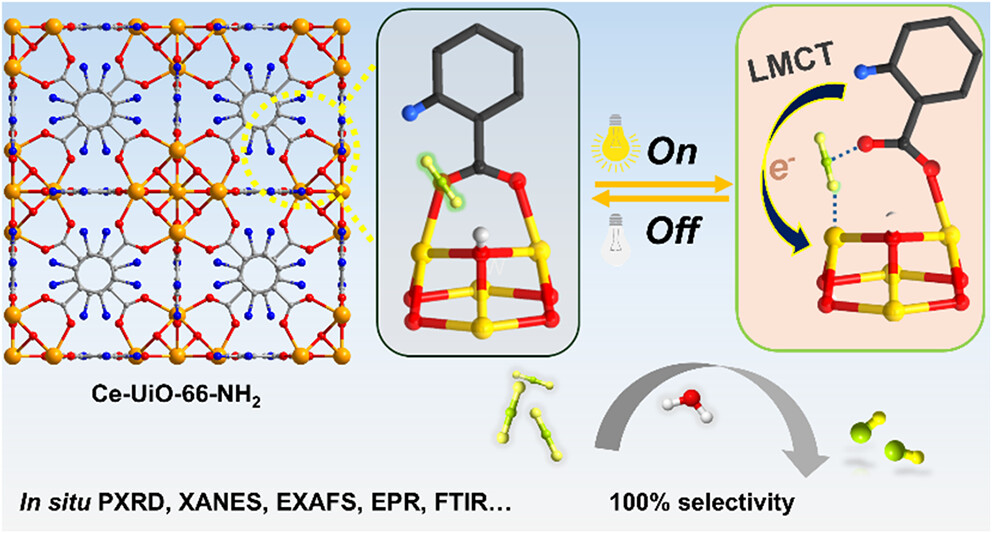
研究背景:
1. 行业问题: 光催化CO₂还原是实现“双碳”目标的关键技术,但面临三大挑战:一是产物选择性低,常伴随析氢副反应(H₂),降低碳效率且产物价值低(合成气价值远低于纯CO);二是传统体系依赖昂贵的牺牲剂(如三乙醇胺)来消耗光生空穴,无法实现真正的水氧化闭环;三是缺乏能在温和条件下高效活化惰性CO₂分子的非贵金属催化剂。
2. 现有方案: 学者们尝试利用金属酶(如碳酸酐酶)的孤立金属位点理念构建MOF催化剂。虽然Zr基或Ce基UiO-66系列材料稳定性优异,但其饱和配位的金属节点难以直接结合底物。现有策略多通过预合成缺陷或高温活化引入开放金属位点,但这些位点往往是静态的,缺乏动态响应能力,且难以在无牺牲剂条件下同时驱动CO₂还原与水氧化。
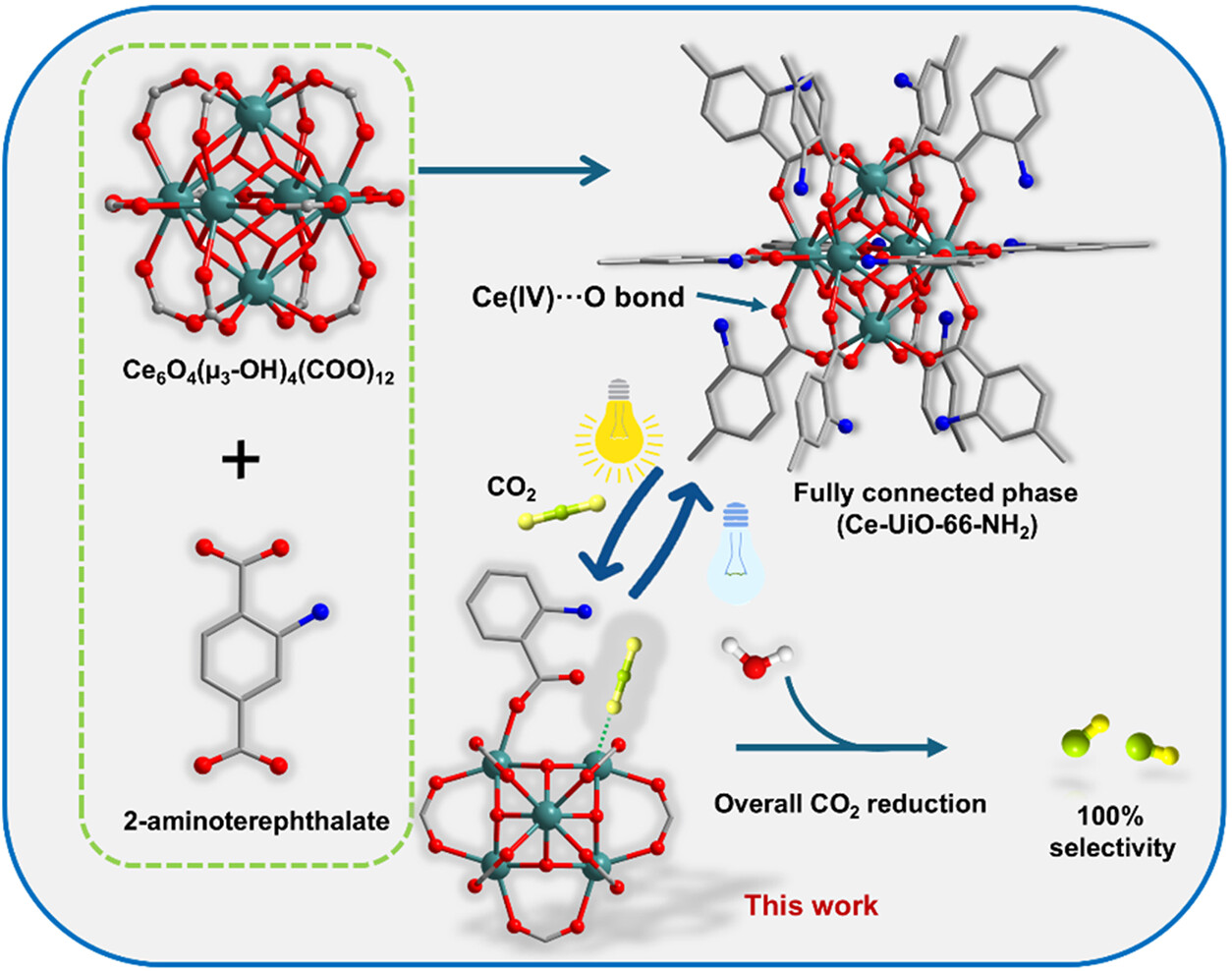
3. 本文创新: 作者受金属酶动态调控启发,提出“光诱导瞬态开放位点”策略。利用氨基修饰的链接体增强可见光吸收及LMCT效应,在光照下瞬时还原Ce(IV)至Ce(III),诱导配体解离产生不饱和位点。这种动态生成的Ce(III)位点能特异性捕获并活化CO₂,且在关灯后可逆恢复,完美解决了活性位点生成与结构稳定性的矛盾,实现了无牺牲剂的全反应循环。
实验部分:
1. 材料合成与对比: 采用溶剂热法合成同构的Ce-UiO-66-NH₂(含氨基)与Ce-UiO-66-H(无氨基)。PXRD证实两者均为高结晶度UiO-66结构。实验发现氨基的引入显著改变了材料的光学性质,使其吸收边红移至可见光区。
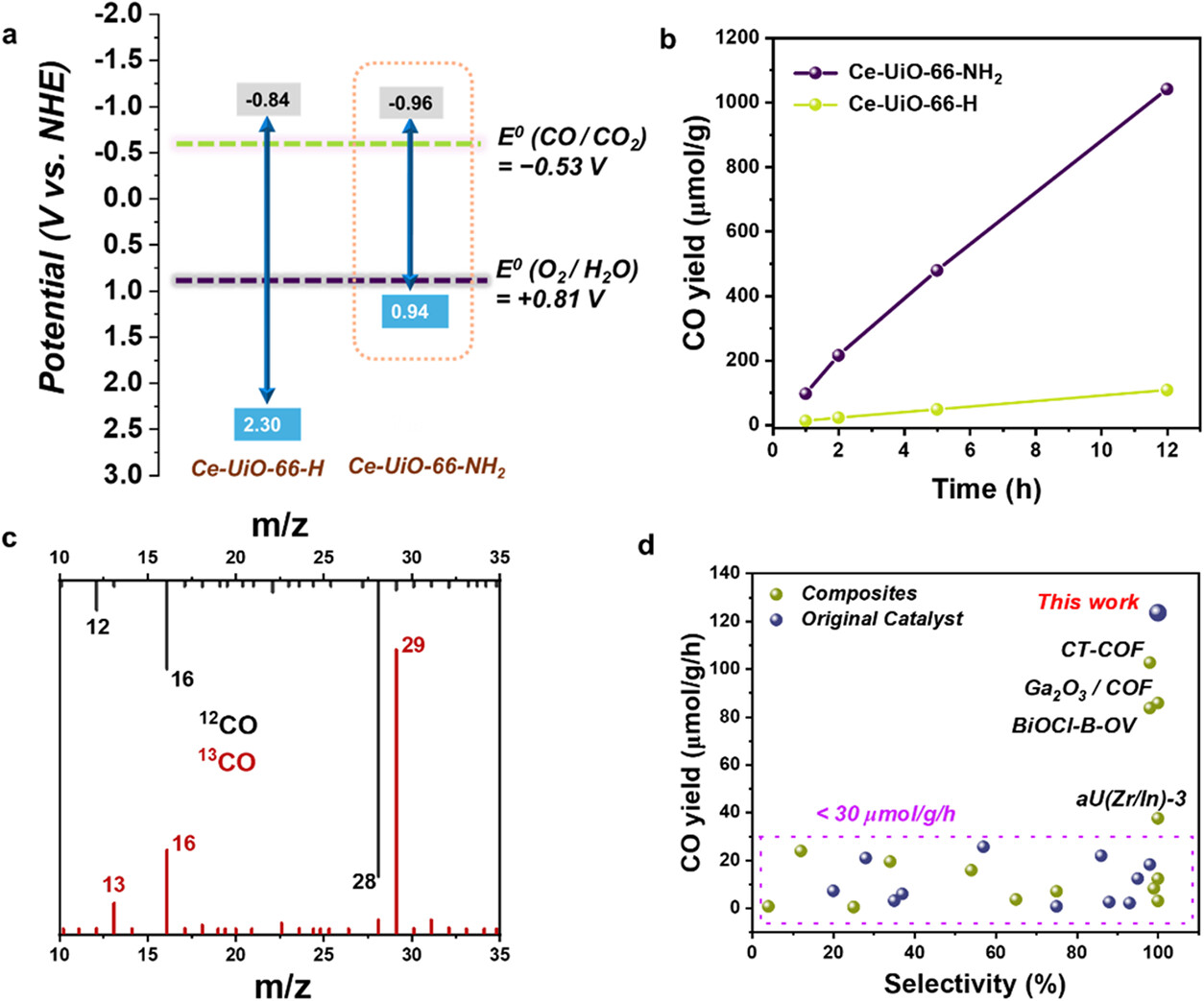
2. 光催化性能测试: 在可见光(Xe灯)照射下,以CO₂和H₂O为原料进行气相光催化反应。结果显示,Ce-UiO-66-NH₂在12小时内持续产生CO,速率为126 μmol·g⁻¹·h⁻¹,且未检测到H₂或其他碳氢产物,选择性达100%。相比之下,Ce-UiO-66-H的活性极低(约5 μmol·g⁻¹·h⁻¹),证明氨基对光活性的决定性作用。
3. 同位素示踪与循环实验: 使用¹³CO₂进行同位素标记实验,质谱检测确认生成的CO完全来源于CO₂而非有机链接体分解。循环测试表明,材料在多次反应后结晶度保持完整,活性无明显衰减,展现了优异的稳定性。
4. 可逆结合验证: 设计了光照开关实验。在光照下通入CO₂观察到明显的化学吸附特征,关闭光源后谱图部分恢复,加热真空处理后可完全恢复至初始状态,证实了CO₂结合的可逆性及光控特性。
表征分析:
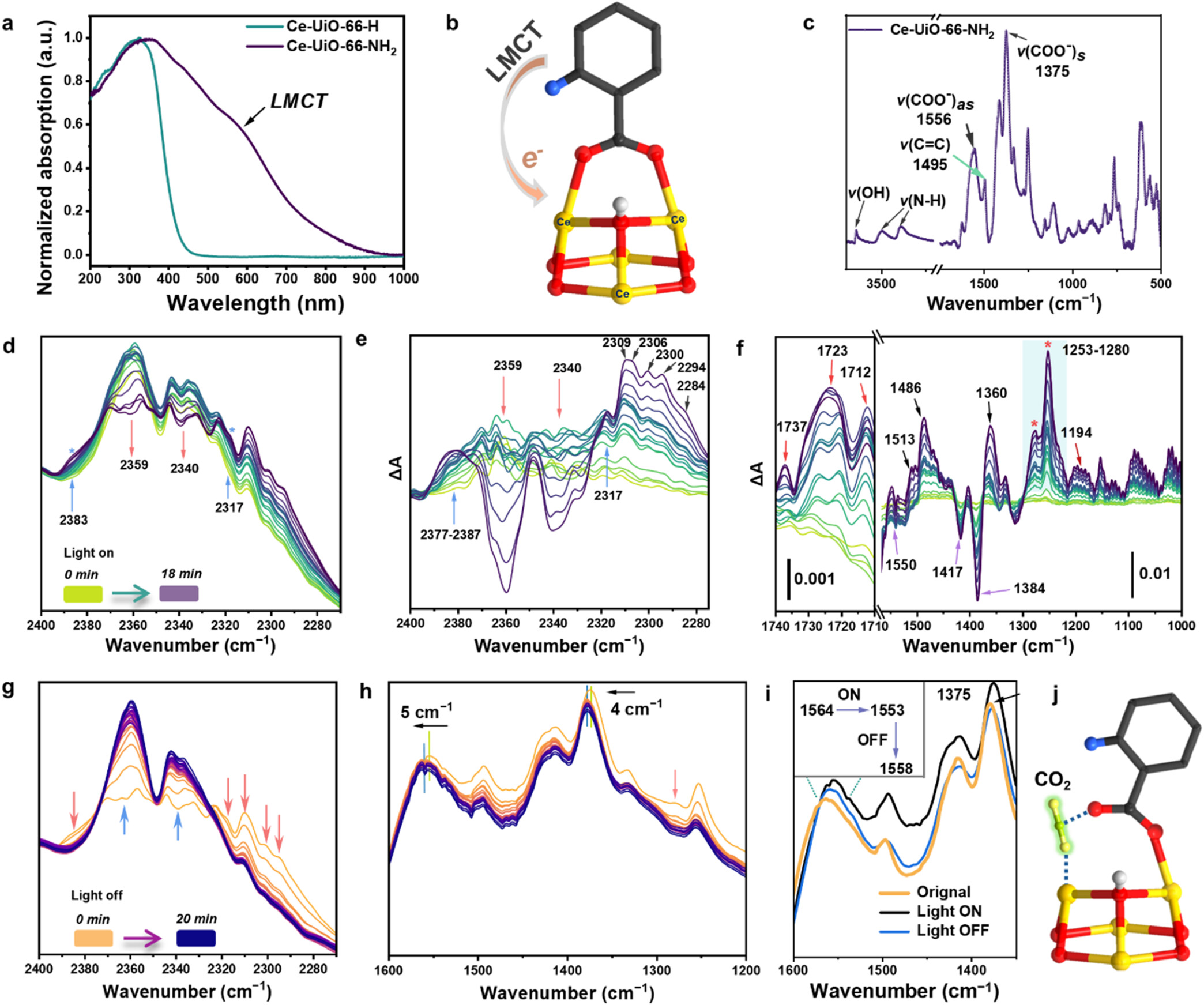
1. 结构与孔隙表征: Ce-UiO-66-NH₂比表面积为624 m²/g,低于Ce-UiO-66-H的1050 m²/g,但其在低压区对CO₂吸附量更高且存在滞后环,暗示氨基增强了主客体相互作用。同步辐射XRD精修表明,CO₂在未光照下主要与μ₃-OH作用(距离2.38 Å),而光照下相互作用显著增强。
2. 光学与能带结构:
1) UV-vis光谱显示Ce-UiO-66-NH₂带隙窄至1.90 eV(Ce-UiO-66-H为3.14 eV)。Mott-Schottky测试确定其导带位置为-0.96 V (vs. NHE),足以驱动CO₂/CO还原(-0.53 V),价带位置满足水氧化需求。
2) 瞬态吸收光谱(TAS)测得Ce-UiO-66-NH₂的LMCT激发态寿命长达218 ns,远优于同类MOF(如Zr-UiO-66-NH₂仅0.36 ns),且在CO₂存在下寿命猝灭至82 ns,证实CO₂是有效的电子受体。
3. 原位光谱监测:
1) 原位FTIR显示,光照下出现2317 cm⁻¹及1275 cm⁻¹新峰,归属为化学吸附活化的CO₂物种;羧酸根伸缩振动红移及1723 cm⁻¹酮式峰的出现,佐证了Ce-O键的断裂与重组。
2) 原位XANES/EXAFS揭示光照下Ce价态由+4向+3动态转变,并出现2.36 Å的新Ce-O键长,对应CO₂配位。
3) EPR检测到光照下生成的顺磁性Ce(III)信号(g值特征)及O₂自由基信号,证实了水氧化过程。
机理分析:
1. 光诱导动态位点生成:
1) 可见光激发引发强烈的配体到金属电荷转移(LMCT),电子从氨基苯二甲酸链接体转移至Ce(IV)节点,将其瞬时还原为Ce(III)。
2) 由于Ce(III)离子半径较大且配位偏好不同,导致原有的Ce-O(羧酸)键减弱甚至断裂,从而在饱和的[Ce₆]簇上动态释放出开放的配位位点。
2. 特异性结合与活化模式:
1) 生成的瞬态Ce(III)位点作为路易斯酸,特异性捕获CO₂分子。不同于传统的端基配位,CO₂在此处以独特的μ-(η1-O)(η1-C)桥连模式结合,即碳原子与Ce(III)结合,一个氧原子与邻近的羧酸氧相互作用。
2) 这种结合模式显著拉长了C=O键,降低了还原能垒,同时空间位阻效应抑制了质子的接近,从而完全抑制了析氢副反应,实现100%的CO选择性。
3. 氧化还原循环: 光生空穴留在有机链接体上,驱动附近的水分子氧化生成O₂和质子,完成电荷平衡。随着CO₂被还原脱附,Ce(III)重新氧化为Ce(IV),羧酸配体重新配位,材料恢复至初始闭合状态,等待下一次光激发循环。
总结:
1. 本文成功构建了首例通过光诱导产生瞬态开放Ce(III)位点的MOF光催化剂,在 Ce-UiO-66-NH2 中,可见光诱导的 LMCT 和电荷分离能够激活 Ce (III) 位点,实现 CO2 的可逆配位。实现了无共催化剂和牺牲剂条件下高选择性(100%)、高效率(126 μmol·g⁻¹·h⁻¹)的CO₂至CO转化。
2. 本研究巧妙地将生物酶的动态活性中心概念引入多孔材料,为设计下一代智能光催化剂提供了全新的视角。
文章标题: Light-Induced Binding and Reduction of CO2 over Transient Open Ce(III) Sites in a Metal–Organic Framework
文章作者: Shan Dai, Xiangdi Zeng, Benjamin J. Moore, et al., Martin Schröder*, Sihai Yang*
DOI: 10.1021/jacs.5c20721
原文链接: https://pubs.acs.org/doi/10.1021/jacs.5c20721
本文为科研用户原创分享,用于学术宣传交流,具体细节请查阅原文。如有错误、侵权,请联系修改删除,未经允许不得复制转载。


 购销咨询
购销咨询 
