首页 >
行业动态 > 【单原子催化剂】质子-电子协同转移过程中甲烷高选择性直接光催化氧化制甲酸
【单原子催化剂】质子-电子协同转移过程中甲烷高选择性直接光催化氧化制甲酸
摘要:
中国科学技术大学刘东\熊宇杰老师等报道的本篇文章(J. Am. Chem. Soc. 2025, 147, 3, 2444–2454)中报道了一种基于WO₃的光催化剂,通过调节Pt活性位点的尺寸效应,实现了甲烷(CH₄)高效选择性转化为甲酸(HCOOH)。研究发现,负载Pt纳米颗粒(PtNPs)的WO₃催化剂在HCOOH的生成率、选择性和稳定性方面均优于未负载Pt的样品以及负载Pt单原子(Pt1)的WO₃催化剂。最佳的PtNPs-WO₃催化剂在光照条件下实现了17.7 mmol g⁻¹的HCOOH转化率,选择性达到84%,并且在48小时的耐久性测试中保持了良好的稳定性。机制研究表明,O₂质子化生成羟基自由基(•OH)是HCOOH产率的限制步骤。Pt纳米颗粒能够促进电子转移并促进O₂解离,通过质子耦合电子转移(PCET)过程生成羟基自由基,从而为•OH自由基的形成提供足够的质子,降低其形成能垒,进而促进CH₄的活化。此外,Pt纳米颗粒还调节了含氧化烃中间体的吸附,提高了反应的选择性。本工作推进了对甲烷转化催化剂设计的理解以及对复杂反应路径的有效调控。

研究背景:
1)甲烷(CH₄)是天然气和生物气的主要成分,也是一种重要的温室气体。将CH₄转化为高附加值化学品对于可持续发展具有重要意义。
直接将CH₄氧化为甲酸(HCOOH)具有挑战性,因为CH₄的C−H键化学惰性强,且需要多个质子和电子参与反应。
光催化转化CH₄为液体产物的研究中,大多数催化剂只能将CH₄氧化为甲醇、甲醛或非选择性的含氧化合物,而选择性转化为HCOOH的研究较少。
2)以往的研究主要集中在通过光催化方法将CH₄转化为液体产物,但大多数研究中HCOOH的选择性较低。
一些研究通过引入贵金属(如Pd)或使用复合催化剂(如C₃N₄-TiO₂)来提高CH₄的转化效率,但这些方法在选择性和稳定性方面仍存在不足。
3)作者设计了基于WO₃的光催化剂,并通过调节Pt活性位点的尺寸(纳米颗粒与单原子)来优化催化性能。
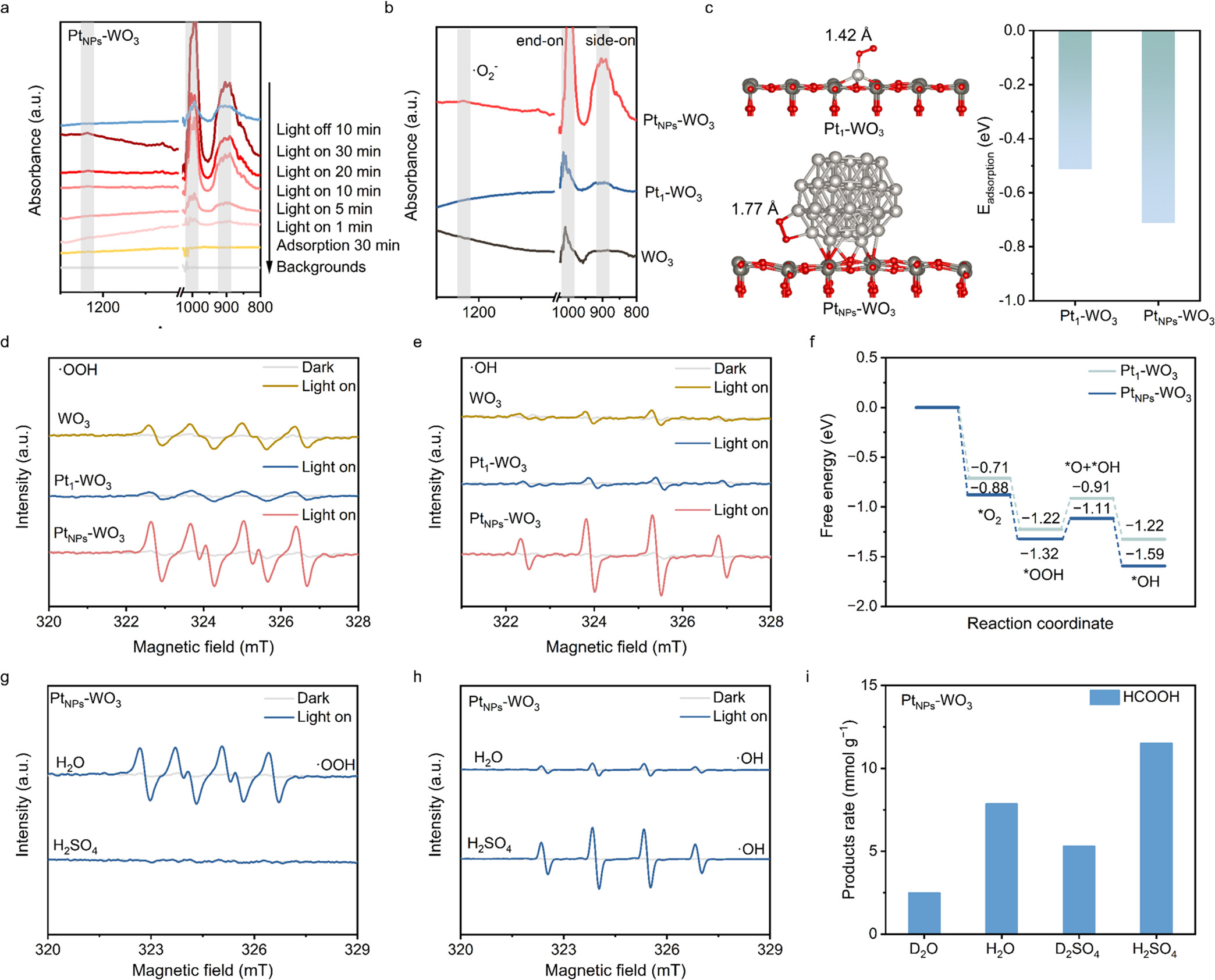
通过实验和理论计算,发现Pt纳米颗粒能够促进O₂的吸附和解离,通过PCET过程生成更多的羟基自由基(•OH),从而提高CH₄的活化效率。
在酸性环境中,质子浓度的增加进一步促进了O₂的活化,提高了HCOOH的选择性和产率。
通过调节Pt纳米颗粒的尺寸,优化了含氧化烃中间体的吸附,提高了HCOOH的选择性。
实验部分:
1)WO₃纳米片的制备:
称取329 mg Na₂WO₄·2H₂O和288 mg柠檬酸,将其溶解在30 mL超纯水中。
向上述溶液中滴加3 mL 6 M的HCl溶液,搅拌均匀。
将混合溶液转移到聚四氟乙烯内衬的反应釜中,在120°C下反应24小时。
反应结束后,将产物通过离心分离,用去离子水多次洗涤,最后在60°C真空干燥箱中干燥。
将干燥后的样品研磨后,在马弗炉中400°C下焙烧2小时,得到WO₃纳米片。
2)PtNPs-WO₃的制备:
称取80 mg WO₃纳米片,将其分散在60 mL超纯水中,超声分散均匀。
向上述分散液中加入120 µL氨水溶液,然后加入0.8 mL H₂PtCl₆·2H₂O溶液(7.5 mg/mL),搅拌均匀后稀释至3 mL。
将400 µL上述溶液滴加到WO₃分散液中,剧烈搅拌10小时。
将混合溶液转移到冰水浴中,然后加入200 µL 0.1 M的NaBH₄溶液,搅拌30分钟。
反应结束后,通过离心分离产物,用去离子水多次洗涤,最后在60°C真空干燥箱中干燥,得到PtNPs-WO₃样品。
3)Pt1-WO₃的制备:
称取80 mg WO₃纳米片,将其分散在50 mL超纯水中,超声分散均匀。
通过注射泵以66.6 µL/min的速度将2 mL Pt(NH₃)₄(NO₃)₂溶液(0.794 mg/mL)缓慢注入WO₃悬浮液中,剧烈搅拌5小时。
反应结束后,通过离心分离产物,用去离子水多次洗涤,最后在60°C真空干燥箱中干燥。
将干燥后的样品在马弗炉中300°C下焙烧2小时,得到Pt1-WO₃样品。
4)光催化CH₄转化实验:
在室温下,将10 mg催化剂分散在120 mL水中,放入石英反应器中。
通入高纯氧气(99.99%)20分钟,排出反应器中的空气,使氧气压力达到1 bar。
在氧气压力达到1 bar后,注入高纯CH₄(99.99%),使总压力达到20 bar。
使用365 nm LED灯(60 mW/cm²)照射反应器,同时持续搅拌。
在不同时间间隔取样,通过气相色谱(GC)检测气态产物,通过核磁共振(NMR)检测液态产物。
分析测试:
1)粉末X射线衍射(XRD):
使用Philips X’Pert Pro Super X射线衍射仪,Cu Kα辐射(λ = 1.54178 Å)。
测试结果表明,WO₃的晶体结构为单斜相,与JCPDS No. 83-0951一致。Pt1-WO₃和PtNPs-WO₃的XRD图谱与WO₃基本一致,未观察到金属Pt的特征峰,说明Pt以高度分散的形式存在于WO₃表面。
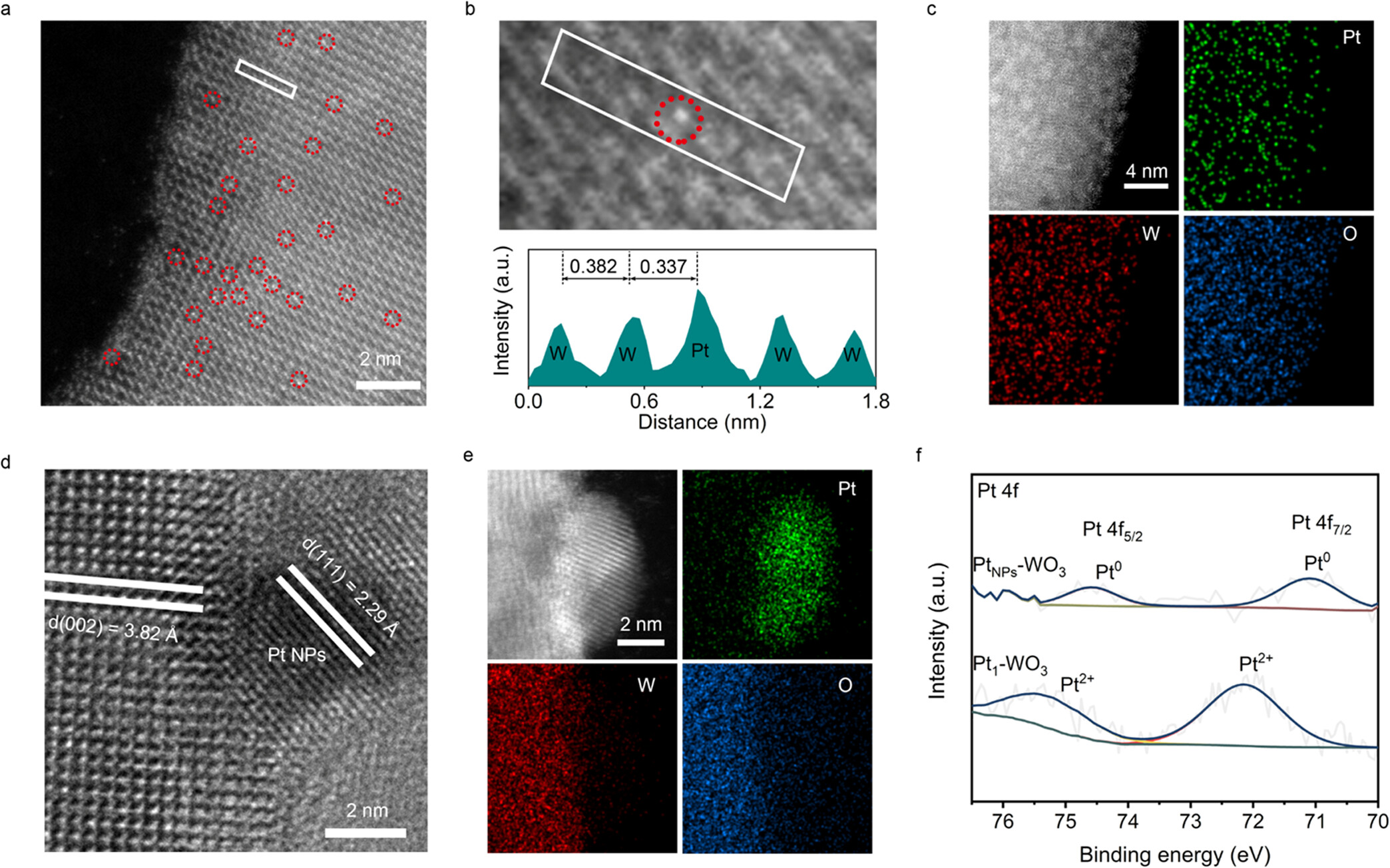
2)透射电子显微镜(TEM):
使用FEI Talos F200X场发射高分辨透射电子显微镜,加速电压为200 kV。
Pt1-WO₃的HAADF-STEM图像显示Pt单原子均匀分散在WO₃纳米片表面,未形成纳米颗粒或团簇。
PtNPs-WO₃的TEM图像显示Pt纳米颗粒均匀分散在WO₃表面,尺寸约为6 nm。
3)X射线光电子能谱(XPS):
使用Thermo Scientific Escalab 250Xi X射线光电子能谱仪,非单色化Al Kα X射线(1486.6 eV)。
PtNPs-WO₃的Pt 4f峰位于71.1 eV(4f₇/₂)和74.5 eV(4f₅/₂),表明Pt为金属态(Pt⁰)。
Pt1-WO₃的Pt 4f峰位于72.1 eV(4f₇/₂)和75.4 eV(4f₅/₂),表明Pt为正电荷态(Ptδ⁺,δ ≈ 2),说明Pt与WO₃形成了Pt−O键。
4)紫外-可见漫反射光谱(UV-Vis DRS):
使用Shimadzu SolidSpec-3700光谱光度计,在200–800 nm范围内记录光谱。
测试结果显示,PtNPs-WO₃和Pt1-WO₃的吸收边与WO₃基本一致,说明Pt的引入未显著改变催化剂的光吸收性能。
5)电子顺磁共振(EPR):
使用JEOL JES-FA200电子自旋共振光谱仪(9.062 GHz)。
在H₂O溶液中,PtNPs-WO₃生成的羟基自由基(•OH)信号强度显著高于Pt1-WO₃和WO₃,表明Pt纳米颗粒能够高效生成•OH自由基。
在CH₃OH溶液中,PtNPs-WO₃生成的超氧自由基(•OOH)信号强度也显著高于Pt1-WO₃和WO₃,进一步证实了Pt纳米颗粒在自由基生成中的重要作用。
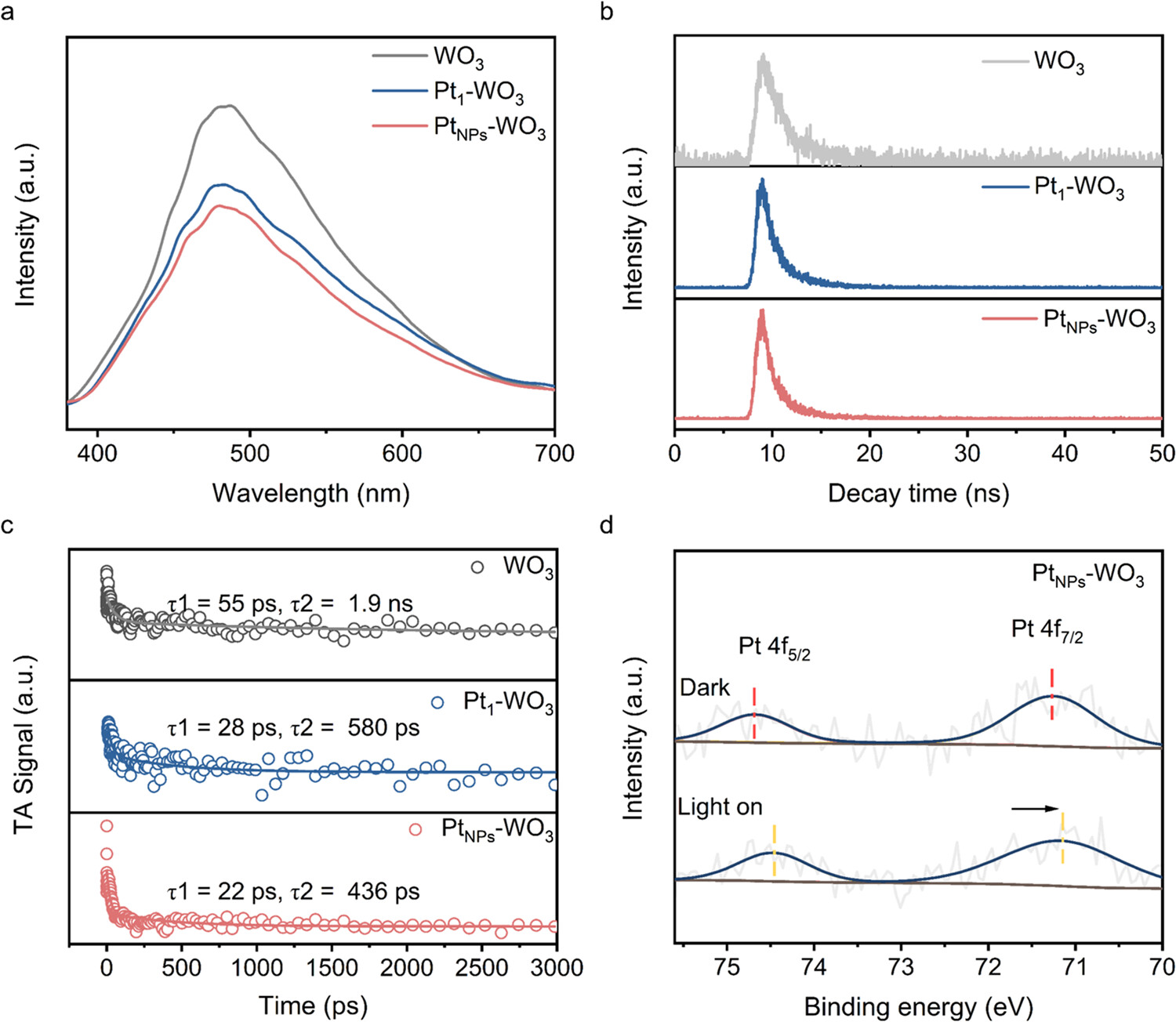
6)光致发光(PL)和时间分辨光致发光(TRPL):
使用Horiba Fluorolog-3荧光光谱仪记录PL光谱。
PtNPs-WO₃的PL光谱在480 nm处的荧光强度低于Pt1-WO₃和WO₃,表明PtNPs-WO₃中光生载流子的复合受到抑制。
TRPL光谱显示,PtNPs-WO₃的平均寿命为1.94 ns,短于Pt1-WO₃(2.18 ns)和WO₃(4.89 ns),说明Pt纳米颗粒促进了光生载流子的分离。
7)原位近常压X射线光电子能谱(NAP-XPS):
使用SPECS NAP-XPS设备,在真空和黑暗条件下记录光谱,然后在365 nm LED光照下记录光谱。
测试结果显示,光照下Pt 4f峰向低结合能方向移动,表明Pt纳米颗粒被光生电子还原,进一步证实了Pt纳米颗粒在光生载流子分离中的作用。
8)甲烷温度程序脱附(CH₄-TPD):
使用Micromeritics AutoChem II 2920仪器进行CH₄-TPD实验。
PtNPs-WO₃在140–170°C和280–360°C处观察到物理吸附和化学吸附峰,表明PtNPs-WO₃对CH₄的吸附能力最强。
9)氧气温度程序脱附(O₂-TPD):
使用Micromeritics AutoChem II 2920仪器进行O₂-TPD实验。
PtNPs-WO₃在240–280°C处观察到一个宽峰,表明PtNPs-WO₃对O₂的吸附能力最强,且在光照下O₂的吸附能力进一步增强。
10)原位漫反射红外傅里叶变换光谱(DRIFTS):
使用Bruker IFS 66 v傅里叶变换光谱仪,配备Harrick漫反射附件,在国家同步辐射实验室(NSRL)的BL01B光束线上进行。
在光照下,PtNPs-WO₃在1011 cm⁻¹(端式吸附O₂)和901 cm⁻¹(侧式吸附O₂)处观察到O₂吸附峰,表明PtNPs-WO₃能够高效吸附和活
总结:
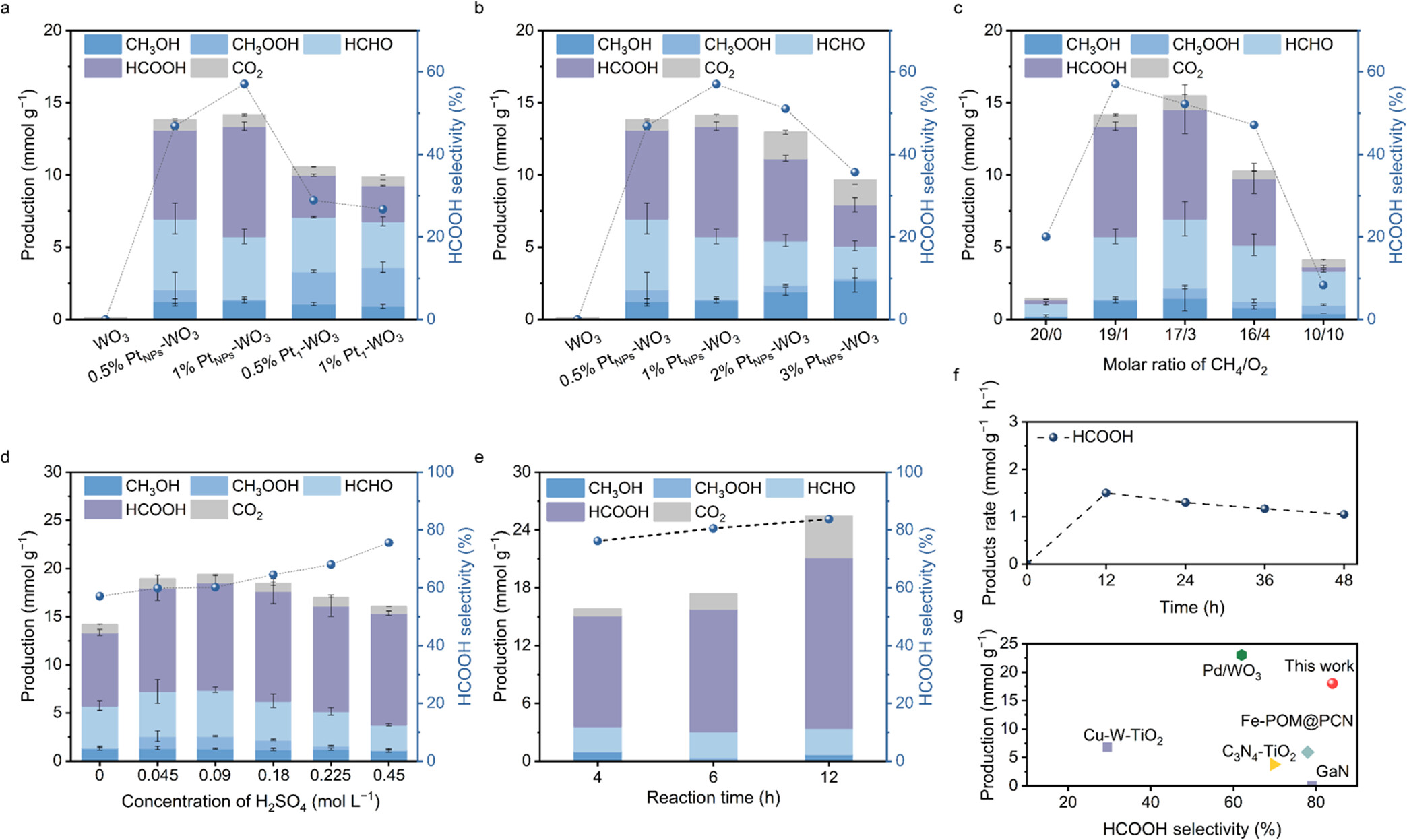
1)本文通过设计基于WO₃的光催化剂,并调节Pt活性位点的尺寸,实现了CH₄高效选择性转化为HCOOH。Pt纳米颗粒的引入显著提高了光生载流子的分离效率,促进了O₂的吸附和解离,通过PCET过程生成更多的•OH自由基,从而提高了CH₄的活化效率。
2)在酸性环境中,质子浓度的增加进一步促进了O₂的活化,提高了HCOOH的选择性和产率。此外,Pt纳米颗粒还优化了含氧化烃中间体的吸附,提高了HCOOH的选择性。最佳的PtNPs-WO₃催化剂在光照条件下实现了17.7 mmol g⁻¹的HCOOH转化率,选择性达到84%,并且在48小时的耐久性测试中保持了良好的稳定性。
展望:
本文的研究成果为甲烷光催化转化提供了重要的理论和实验基础。未来的研究可以进一步探索以下方向:
催化剂的长期稳定性:虽然本文的催化剂在48小时内表现出良好的稳定性,但需要进一步研究其在更长时间内的性能变化。
反应机理的深入研究:通过更详细的原位表征技术(如原位XAS、原位拉曼光谱等)进一步揭示反应过程中中间体的形成和转化机制。
催化剂的规模化制备:探索催化剂的大规模制备方法,以满足工业应用的需求。
反应条件的优化:进一步研究反应条件(如光照强度、反应温度、气体压力等)对催化性能的影响,以实现更高的转化效率和选择性。
环境影响评估:评估该光催化过程对环境的影响,特别是酸性条件下的生态安全性。
Direct Photocatalytic Oxidation of Methane to Formic Acid with High Selectivity via a Concerted Proton−Electron Transfer Process
文章作者:Guangyao Zhai, Siyuan Yang, Yihong Chen, Junchi Xu, Shenghe Si, Honggang Zhang, Yuanyuan Liu, Jun Ma, Xiao Sun, Weixin Huang, Chao Gao, Dong Liu,* and Yujie Xiong*
DOI:https://doi.org/10.1021/jacs.4c12758
文章链接:https://pubs.acs.org/doi/10.1021/jacs.4c12758
本文为科研用户原创分享上传用于学术宣传交流,具体内容请查阅上述论文,如有错误、侵权等请联系修改、删除。未经允许第三方不得复制转载。


 购销咨询
购销咨询 
